
- Beiträge: 1763
Sidebar
- Forum
- PH-Forum
- Pulmonale Hypertonie, Lungenhochdruck - Kinder und Jugendliche
- Übersichtsartikel, PH bei Kindern
Übersichtsartikel, PH bei Kindern
28 Dez 2023 15:29 #1979
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Übersichtsartikel, PH bei Kindern wurde erstellt von danny
erj.ersjournals.com/content/53/1/1801916AbstraktDie pädiatrische pulmonale arterielle Hypertonie (PAH) weist gemeinsame Merkmale der Erkrankung bei Erwachsenen auf, ist jedoch mit mehreren zusätzlichen Störungen und Herausforderungen verbunden, die einzigartige Ansätze erfordern. In diesem Artikel werden aktuelle Fortschritte, aktuelle Herausforderungen und unterschiedliche Ansätze für die Versorgung von Kindern mit PAH erörtert, wie sie von der Pediatric Task Force des 6. Weltsymposiums zur pulmonalen Hypertonie vorgestellt wurden. Wir stellen Aktualisierungen der aktuellen Definition, Epidemiologie, Klassifizierung, Diagnostik und Behandlung der pädiatrischen PAH bereit und identifizieren kritische Wissenslücken. Mehrere Merkmale der pädiatrischen PAH werden hervorgehoben, darunter die Bedeutung der neonatalen PAH, insbesondere bei Frühgeborenen mit sich entwickelnden Lungenerkrankungen, und neue genetische Ursachen der pädiatrischen PAH. Der Einsatz der Herzkatheterisierung als diagnostische Modalität und hämodynamische Definitionen von PAH, einschließlich akuter Vasoreaktivität, werden behandelt. Es werden Aktualisierungen zu Fragen im Zusammenhang mit der Nützlichkeit des vorherigen Klassifizierungssystems bereitgestellt, um pädiatriespezifische Ätiologien und Ansätze zur medizinischen und interventionellen Behandlung von PAH, einschließlich des Potts-Shunts, widerzuspiegeln. Obwohl es nach wie vor an klinischen Studiendaten für den Einsatz einer PAH-zielgerichteten Therapie mangelt, verbessern neue Daten die Identifizierung geeigneter Ziele für eine zielgerichtete Therapie bei Kindern. Solche Daten werden wahrscheinlich das Design zukünftiger klinischer Studien verbessern, um die Ergebnisse bei pädiatrischer PAH zu verbessern.Tweetbare Zusammenfassung @ERSpublications
Klicken Sie hier, um zu twittern
Stand der Technik und Zukunftsperspektiven bei pädiatrischer pulmonaler Hypertonie mit besonderem Schwerpunkt auf Klassifizierung, Diagnose und Behandlung
ow.ly/uVPo30mksOj
EinführungPulmonale Hypertonie (PH) bei Kindern ist mit verschiedenen Erkrankungen verbunden, die in jedem Alter auftreten können. Die Verteilung der Ätiologien bei pädiatrischer PH unterscheidet sich deutlich von der bei Erwachsenen, wobei bei Kindern die idiopathische pulmonale arterielle Hypertonie (IPAH), die pulmonale arterielle Hypertonie (PAH) in Verbindung mit angeborenen Herzfehlern (PAH-KHK) und entwicklungsbedingte Lungenerkrankungen häufiger vorkommen . Unterschiede in Ätiologie, Präsentation und Ergebnissen erfordern einen einzigartigen Ansatz bei Kindern. Die Behandlung von Kindern bleibt eine Herausforderung, da Behandlungen seit langem auf evidenzbasierten Erwachsenenstudien und der klinischen Erfahrung pädiatrischer Experten basieren. Obwohl es noch an Daten zu Wirksamkeit, Formulierung, Pharmakokinetik, optimaler Dosierung und Behandlungsstrategien mangelt, zeichnen sich Daten ab, die die Definition geeigneter Behandlungsziele und eine zielgerichtete Therapie bei Kindern ermöglichen. Dennoch werden Kinder mit PAH derzeit mit gezielten PAH-Medikamenten erfolgreich behandelt. Wir bieten einen Überblick über aktuelle Aktualisierungen der aktuellen Definition, Epidemiologie, Klassifizierung, Diagnostik und Behandlung von PAH bei Kindern und ermitteln den aktuellen Bedarf auf der Grundlage von Diskussionen und Empfehlungen der Pediatric Task Force des 6. Weltsymposiums zu pulmonaler Hypertonie (WSPH) in Nizza, Frankreich (2018).DefinitionenHistorisch gesehen war die Definition von PH bei Kindern dieselbe wie bei Erwachsenen, dh mittlerer pulmonaler arterieller Druck (mPAP) ≥25 mmHg. Im normalen fetalen Kreislauf ähnelt der PAP dem systemischen Druck und sinkt nach der Geburt schnell ab, um im Alter von 2–3 Monaten ähnliche Werte wie beim Erwachsenen zu erreichen. Aufgrund der Variabilität der pulmonalen Hämodynamik während des postnatalen Übergangs wurde ein pH-Wert bei Kindern als mPAP ≥ 25 mmHg nach einem Alter von 3 Monaten definiert. Bei pädiatrischer PH, insbesondere in Verbindung mit KHK, wird empfohlen, den pulmonalen Gefäßwiderstand (PVR) in Abhängigkeit von der Körperoberfläche (PVRI) zu verwenden, um das Vorliegen einer pulmonalen Gefäßerkrankung (PVD) gemäß Definition durch PVRI ≥3 zu beurteilen WU·m 2 .Das 6. WSPH schlug vor, die Definition für PH bei Erwachsenen als mPAP > 20 mmHg zu ändern und PVR ≥ 3 WU einzubeziehen, um präkapilläre PH zu identifizieren . Ob die gleichen Befunde einer hohen normalen oder „grenzwertigen PH“ mit mPAP von 21–24 mmHg in Untergruppen erwachsener PH wie Sklerodermie, chronisch obstruktiver Lungenerkrankung und interstitieller Lungenerkrankung vorliegen oder nicht, ist ein Risikofaktor für die Entwicklung von PAH und damit verbundenen Morbiditäten bei Kindern wie bei Erwachsenen bedarf weiterer Untersuchungen [
1
]. Um jedoch eine universelle Sprache zu sprechen und den Übergang von der pädiatrischen PH-Versorgung zur erwachsenen PH-Versorgung zu erleichtern, hat sich die Pädiatrische Task Force dafür entschieden, der neu vorgeschlagenen PH-Definition für Erwachsene zu folgen, und ermutigt zu weiteren Untersuchungen dieser Patienten. Der vorgeschlagene Einsatz von PVR zur Beurteilung der PVD am 6. WSPH war bereits zuvor in die hämodynamische Beurteilung der PH bei Kindern einbezogen worden. Die Pediatric Task Force hat die Notwendigkeit einer Indexierung der PVR bei Kindern weiter bekräftigt.VasoreaktivitätBei IPAH/erblicher PAH (HPAH) wird ein akuter Vasodilatatortest (AVT) empfohlen, um Patienten zu identifizieren, die wahrscheinlich eine gute Langzeitprognose haben, wenn sie mit einer Langzeittherapie mit Kalziumkanalblockern (CCB) behandelt werden. In der jüngeren Vergangenheit ging man davon aus, dass eine positive Vasoreaktivität bei Kindern mit IPAH im Vergleich zu Erwachsenen häufiger vorkommt und dass für Kinder spezifische Ansprechkriterien angegeben wurden. Die Verwendung derselben Kriterien für erwachsene und pädiatrische Probanden mit IPAH/HPAH zeigte jedoch, dass der Anteil der AVT-Responder in beiden Altersgruppen ähnlich war [
2
]. Eine kürzlich durchgeführte große Studie mit 382 Patienten zeigte eine erhebliche Diskrepanz in der Art und Weise, wie AVT bei Kindern durchgeführt und interpretiert wird, und dass eine Standardisierung erforderlich ist [
2
]. Wie bei Erwachsenen ist inhaliertes Stickstoffmonoxid mit 10–80 ppm das bevorzugte Mittel, alternativ können aber auch iv Epoprostenol, iv Adenosin oder inhaliertes Iloprost verwendet werden. Für die letztgenannten Arzneimittel ist die optimale Dosierung bei Kleinkindern jedoch nicht genau definiert. Wie kürzlich berichtet, haben sich die Sitbon-Kriterien für eine positive AVT, definiert durch eine Abnahme des mPAP um mindestens 10 mmHg auf einen Wert von <40 mmHg bei anhaltendem Herzzeitvolumen, die üblicherweise bei IPAH/HPAH bei Erwachsenen verwendet werden, zur Identifizierung von Kindern erwiesen die von der CCB-Therapie nachhaltig profitieren werden [
2
,
3
] . Eine positive AVT bei Probanden mit mPAP <40 mmHg zu Studienbeginn ist durch einen Abfall von mindestens 10 mmHg ohne Abfall des Herzzeitvolumens definiert. Basierend auf diesen Daten wird empfohlen, die Sitbon-Kriterien für AVT bei Kindern zu verwenden. Da gezeigt wurde, dass nur bei der Hälfte der erwachsenen Patienten, die auf die CCB-Therapie reagierten, eine langfristige hämodynamische und klinische Verbesserung zu verzeichnen war, ist eine engmaschige Langzeitbeobachtung erforderlich.Kann die AVT die Funktionsfähigkeit vorhersagen, wenn der Ruhe-PAP und der PVR bei Kindern mit KHK und offenen systemisch-pulmonalen Shunts erhöht sind?Bei KHK-assoziierter PH wird eine AVT häufig aus anderen Gründen durchgeführt als zur Bestimmung des potenziellen Nutzens einer CCB-Therapie und zur Vorhersage des Ergebnisses, wie bei IPAH/HPAH gezeigt. AVT wird auch verwendet, um bei Patienten mit PAH-KHK zwischen reversibler und progressiver PAH und damit potenzieller Operabilität zu unterscheiden [
4
]. Spezifische Kriterien zur Definition einer positiven AVT-Reaktion oder spezifische hämodynamische Ziele, die eine Umkehr der PAH und eine gute Langzeitprognose nach einer chirurgischen Korrektur vorhersagen, fehlen jedoch weiterhin. Tatsächlich wurde gezeigt, dass neben der hämodynamischen Reaktion auf AVT auch andere Faktoren mit der Umkehrung der PAH nach einer chirurgischen Reparatur in Zusammenhang stehen, darunter Alter, Art der Herzläsion, Komorbiditäten, Ruhe- und Belastungssättigung sowie klinische Vorgeschichte. Da keine belastbaren Daten zu hämodynamischen Prädiktoren vorliegen, schlagen die aktuellen Leitlinien Kriterien für die Operabilität einer KHK bei Vorliegen einer PAH vor, die auf Expertenmeinungen basieren. Die Pädiatrische Task Force einigte sich auf allgemeine Leitlinien zur Beurteilung der Operabilität bei KHK-PAH, betont jedoch, dass die langfristigen Auswirkungen des Defektverschlusses bei Vorliegen einer PAH mit erhöhtem PVR unbekannt sind (
Tabelle 1
).
TABELLE 1Leitfaden zur Beurteilung der Operabilität bei pulmonaler arterieller Hypertonie im Zusammenhang mit angeborenen Herzfehlern
Aktualisierungen der pädiatrischen PH-Epidemiologie und -KlassifizierungAktuelle epidemiologische Daten zur pädiatrischen PH stammen hauptsächlich aus Registerkohorten; Infolgedessen werden sie vom Studiendesign, der Logistik und dem Umfang der klinischen Praxis beeinflusst, die der Patientenauswahl für diese Register zugrunde liegt. Geografische Abdeckung, Überweisungsmuster, Einschlusskriterien und Krankheitsdefinitionen können zwischen den Registern unterschiedlich sein, was zu einer potenziellen Selektionsverzerrung führen kann, die sich auf die gemeldeten Inzidenz- und Prävalenzraten auswirken kann. Die geschätzte Inzidenz anhaltender PH in allen Kategorien wurde mit 4–10 Fällen pro Million Kinder und Jahr angegeben, mit einer Prävalenz von 20–40 Fällen pro Million in Europa (Spanien, Niederlande) und 5–8 Fällen pro Million Kindern und Jahr
26–33
pro Million Kinder in den USA [
5–7
]. Eine landesweite epidemiologische Studie in den Niederlanden, die potenzielle Verzerrungen minimierte, indem sie alle Krankenhäuser des Landes einbezog und Krankenhausregister mit lokalen pädiatrischen kardiologischen Datenbanken über einen Zeitraum von 15 Jahren kombinierte, berichtete über eine jährliche Inzidenzrate von PH bei Kindern von 63,7 pro Million Kinder [
6
] . Die Mehrheit der Kinder (2845 von 3262) hatte eine „vorübergehende“ PAH und waren Säuglinge mit entweder persistierendem PH des Neugeborenen (PPHN) oder reparablen Herz-Shunt-Defekten. Von den übrigen Kindern hatten 27 % andere Formen von PAH (IPAH, PAH-CHD, PAH assoziiert mit Bindegewebserkrankung (CTD) und pulmonaler venöser Verschlusskrankheit (PVOD)), während ein erheblicher Anteil (34 %) PH-assoziiert aufwies mit sich entwickelnder Lungenerkrankung, einschließlich bronchopulmonaler Dysplasie (BPD), angeborener Zwerchfellhernie (CDH) und angeborenen Lungengefäßanomalien [
6
].Gruppe 1: PAHGruppe 1.1: IPAH
Die geschätzten
Inzidenzraten für IPAH variieren zwischen 0,47 und 1–2 Fällen pro Million Kinder, mit geschätzten Prävalenzraten zwischen 2,1 und 4,4 Fällen pro Million Kindern [
5–9
].Gruppe 1.2: HPAHWie bei der PAH bei Erwachsenen wurden bei 20–30 % der pädiatrischen sporadischen PAH-Fälle und bei 70–80 % der familiären PAH-Fälle Genmutationen identifiziert, die an der Pathogenese der HPAH beteiligt sind. Dazu gehören bekannte Mutationen wie die in BMPR2 (Bone Morphogenetic Protein Receptor Type 2) und ACVRL1 (Activin Receptor-like Kinase 1).
Im Vergleich zur PAH bei
Erwachsenen unterscheidet sich die genetische Architektur der pädiatrischen PAH jedoch und scheint mit TBX4- und ACVRL1- Mutationen angereichert zu sein [
10–13
]. Ob Mutationsträger bei pädiatrischer PAH einen anderen Phänotyp oder klinischen Verlauf haben als Nichtträger, muss noch nachgewiesen werden [
11
]. Darüber hinaus geht pädiatrische PAH häufig mit Chromosomen- und syndromalen Anomalien einher, bei denen die mechanistische Grundlage für PAH im Allgemeinen ungewiss ist. Eine kürzlich durchgeführte Exomsequenzierungsstudie bei pädiatrischer PAH legt nahe, dass De-novo- Varianten in neuen Genen etwa 19 % der IPAH-Fälle mit pädiatrischem Beginn erklären könnten. Die Prävalenz bekannter PAH-Genmutationen bei PAH-KHK ist umstritten, da in mehreren Studien keine PAH-Mutationen bei diesen Patienten nachgewiesen wurden, während andere Gruppen BMPR2- Mutationen bei Patienten identifiziert haben, die nach Korrektur eines Defekts an PAH erkrankten [
11
,
14
] und a Eine aktuelle Studie zeigt Varianten von SOX17 im Zusammenhang mit PAH-KHK [
15
]. Gentests werden immer noch nicht routinemäßig bei allen pädiatrischen PH durchgeführt, da sie zu erheblichen psychologischen Auswirkungen führen können, insbesondere bei asymptomatischen Personen, insbesondere bei unvollständiger Penetranz. Gentests sollten mit einer genetischen Beratung durch Experten auf diesem Gebiet kombiniert werden, damit Familien vor und nach dem Test über alle Informationen verfügen.Die Genetik der PH und Gentests speziell in der Pädiatrie erfordern weitere Arbeiten und sollten in Expertenzentren mit einer genetischen Beratungsgruppe durchgeführt werden.Gruppe 1.3: Arzneimittel- und toxininduzierte PAHIn der Literatur wurden mehrere Fälle von vorübergehender PAH beschrieben, die nach Absetzen von Diazoxid verschwanden, was darauf hindeutet, dass hyperinsulinämische und hypoglykämische Neugeborene, die mit Diazoxid behandelt werden, eine echokardiographische Überwachung benötigen [
16
]. Die US-amerikanische Food and Drug Administration (FDA) gab 2015 eine Warnung vor Diazoxid und PAH bei Neugeborenen heraus [
17
].Gruppe 1.4: Assoziierte PAHGRUPPE 1.4.1: PAH IM ZUSAMMENHANG MIT CTDPAH-CTD kommt bei Kindern selten vor, aber wenn sie mit PAH einhergeht, kommt es in der Regel schnell zu einer Verschlechterung. PAH-CTD tritt bei
0–4
% der Patienten in PH-Kliniken auf [
5–7
,
18–21
]
. In einer multinationalen Kohorte von 389 Kindern mit systemischer juveniler Arthritis aus dem CARRA-Register wurde bei 16 (4 %) PAH diagnostiziert [
18
]. Eine aktuelle Studie legt nahe, dass PAH innerhalb der ersten zwei Jahre nach der Diagnose bei 2 % einer Kohorte von 850 Kindern mit systemischem Lupus erythematodes mittels Echokardiographie diagnostiziert wurde. Diese Patienten mit PH waren größtenteils asymptomatisch und in einigen Fällen verschwand die PAH oder besserte sich [
19
].GRUPPE 1.4.2: PAH IM ZUSAMMENHANG MIT EINER HIV-INFEKTIONPAH-HIV bei Kindern scheint außerhalb von Endemiegebieten selten zu sein, mit jeweils einem Fall in den spanischen, niederländischen und britischen Registern [
5
,
6
,
22
].GRUPPE 1.4.3: PAH IM ZUSAMMENHANG MIT PORTALER HYPERTONIEPatienten mit Lebererkrankungen leiden an zwei unterschiedlichen pulmonalen Gefäßkomplikationen: dem hepatopulmonalen Syndrom (HPS) und der portopulmonalen Hypertonie (POPH). Während HPS durch einen erhöhten pulmonalen Blutfluss, einen niedrigen PVR und Hypoxämie gekennzeichnet ist, weist POPH eine auffällige pulmonale Gefäßumgestaltung auf, die sich negativ auf das Ergebnis einer orthotopen Lebertransplantation auswirkt [
23
]. POPH scheint bei Kindern selten zu sein, wobei 0–2 % der Fälle in PH-Registern gemeldet werden [
5
,
6
,
22
,
24
].GRUPPE 1.4.4: ANGEBORENE HERZFEHLERGruppe 1.4.4 PAH umfasst Patienten mit einfach operabeler und inoperabler KHK, unterteilt in Patienten mit Eisenmenger-Physiologie, Patienten mit PAH und Links-Rechts-Shunts, Patienten mit PAH, von denen angenommen wird, dass sie mit ihrer KHK in Zusammenhang stehen, und Patienten mit postoperativen/geschlossenen Defekten. Diese Klassifizierung für PAH im Zusammenhang mit kardialen oder arteriellen Shunts hat sich seit der vorherigen WSPH-Klassifizierung von 2013 nicht geändert [
25
]. Vorübergehende PH nach der Reparatur angeborener Herzfehler treten in 21,9 Fällen pro Million auf und sind nach der persistierenden pulmonalen Hypertonie des Neugeborenen eine der häufigsten Formen von PAH bei Kindern [
6
]. Komplexe Herzerkrankungen wurden der Gruppe 5.4 zugeordnet.GRUPPE 1.4.5: BILHARZIOSESchistosomiasis ist in Industrieländern selten und es fehlen Studien zur gezielten PH-Therapie bei Kindern.Gruppe 1.5: PAH-Langzeit-Responder auf CCBsWie bei Erwachsenen kann eine Untergruppe von Kindern mit IPAH identifiziert werden, die positiv auf AVT reagieren und nun als „PAH-Langzeit-Responder auf CCBs“ klassifiziert werden [
25
]. Basierend auf den Sitbon-Kriterien umfasst diese Untergruppe schätzungsweise etwa 8–15 % der Kinder mit IPAH.Gruppe 1.6: PAH mit offensichtlichen Merkmalen einer venösen/kapillaren (PVOD/PCH)-BeteiligungPAH mit offensichtlichen Merkmalen einer venösen/kapillaren Beteiligung (PVOD/pulmonale kapilläre Hämangiomatose (PCH)) scheint bei Kindern selten zu sein. PVOD und/oder PCH wurden in 0,7–2 % der PAH-Fälle in den spanischen, niederländischen und TOPP-Registern diagnostiziert [
5
,
6
,
26
]. Die EIF2AK4- Mutation (eukaryotischer Translationsinitiationsfaktor 2α-Kinase 4) war bei zwei Dritteln der in Frankreich mit PVOD diagnostizierten Kinder vorhanden [
11
].Gruppe 1.7: Anhaltende PH des NeugeborenensyndromsPPHN ist die häufigste Ursache für vorübergehende PAH (30,1 Fälle pro Million Kinder pro Jahr) und kann an Häufigkeit zunehmen [
7
]. In der Zeit vor der inhalativen Stickoxidtherapie trat PPHN bei etwa 2 von 1000 Lebendgeburten auf [
27
]. Im Gegensatz dazu lag die Prävalenz von PPHN zwischen 2003 und 2012 bei 12.954 eingeschriebenen extrem frühgeborenen Säuglingen bei 8,1 % (95 %-KI 7,7–8,6 %), wobei der Trend jährlich zunahm, was teilweise auf die erhöhte Überlebensrate extrem niedrig geborener Säuglinge zurückzuführen war. Geburtsgewicht von Säuglingen und wachsendes Bewusstsein für PPHN bei Frühgeborenen. Der Anteil der Neugeborenen mit PPHN steht im umgekehrten Verhältnis zum Gestationsalter, mit einer Inzidenz von 18,5 % (Bereich 15,2–22,4 %) bei Säuglingen, die in der 22.–24. Woche geboren wurden, verglichen mit 4,4 % (Bereich 3,8–5,2 %) bei Säuglingen, die mit 27 Jahren geboren wurden Wochen [
28
]. Die aktuelle WSPH Pediatric Task Force betonte, dass es sich bei PPHN um ein Syndrom mit mehreren Begleiterkrankungen handelt (
Tabelle 2
). Obwohl PPHN multifaktoriellen Ursprungs ist, zeigen neuere epidemiologische Studien, dass PPHN mit vorgeburtlichen Ereignissen, einschließlich Präeklampsie, Chorioamnionitis und anderen perinatalen Ereignissen, verbunden ist, was zu abnormalem Wachstum und Funktion der Lungengefäße führt und möglicherweise das Risiko einer späteren PAH erhöht Leben [
26
].
TABELLE 2Anhaltende pulmonale Hypertonie des Neugeborenen (PPHN) und damit verbundene Erkrankungen
Gruppe 2: PH aufgrund einer Erkrankung des linken HerzensZu dieser Erkrankung bei Kindern liegen nur sehr wenige epidemiologische Daten vor; Allerdings können eine linksventrikuläre diastolische Dysfunktion und eine beeinträchtigte Myokardleistung in verschiedenen Situationen, einschließlich PPHN, BPD und CDH, zum PH-Schweregrad beitragen. Angeborene Obstruktionen des Ein- und Ausflusses des linken Herzens kommen bei Kindern mit koronarer Herzkrankheit häufig vor, und das Ergebnis hängt von der Ätiologie und dem Stadium der pulmonalen Gefäßentwicklung ab, in dem die Obstruktion auftritt. Eine Pulmonalvenenstenose, die eine sehr schlechte Prognose hat, kann den Verlauf komplizieren und entwickelt sich zu einer wichtigen
Ursache
für eine anhaltende PH, insbesondere bei Frühgeborenen mit BPD [
29–32
].
Tabelle 3
zeigt angeborene postkapilläre obstruktive Läsionen, die im Kindesalter am häufigsten vorkommen und nun der Gruppe 2,4 PH zugeordnet sind
TISCH 3Angeborene postkapilläre obstruktive Läsionen (Gruppe 2.4)
Gruppe 3: PH aufgrund von Lungenerkrankungen und/oder HypoxieGruppe 3.5: Entwicklungsbedingte LungenstörungenDiese Kategorie umfasst einen wichtigen und zunehmend anerkannten Anteil von Kindern mit PH. BPD ist eine häufige Entwicklungsstörung bei Frühgeborenen, die durch eine Beeinträchtigung des alveolären und vaskulären Wachstums und der Reifung gekennzeichnet ist. Aus früheren Registerdaten geht hervor, dass 10–12 % der Kinder mit PH eine assoziierte Lungenerkrankung haben, wobei BPD die häufigste Erkrankung ist [
26
,
33
]. Dies könnte, wie bereits erwähnt, aufgrund der Verzerrung bei der Registrierung in das Register wahrscheinlich eine Unterrepräsentation der Häufigkeit sein. Die niederländische epidemiologische Studie ergab, dass 34 % der Patienten mit anhaltender PH an einer sich entwickelnden Lungenerkrankung leiden. Die Inzidenz und Prävalenz schwerer BPD und PH nehmen mit zunehmender Überlebenszeit von 23–26 Wochen vor der Entbindung zu. In einer prospektiven Studie war PH im Alter von 7 Tagen bei 42 % der Frühgeborenen (Geburtsgewicht 500–1250 g) vorhanden und war bei 14 % mit einem späten PH (im korrigierten Alter von 36 Wochen), einem schlimmeren Schweregrad der BPD und einem längeren Bedarf verbunden für mechanische Beatmung und Krankenhausaufenthalte auf der Neugeborenen-Intensivstation sowie eine höhere Sterblichkeit [
34
,
35
]. Bei Kindern mit Borderline-Persönlichkeitsstörung (BPS) kann PH mit der Zeit durch respiratorische und PH-zielgerichtete medikamentöse Therapie verschwinden; Allerdings bleibt die Morbidität und Mortalität von PH bei BPD-Säuglingen auch im Surfactant-Zeitalter hoch. Jüngste Metaanalysen ergaben, dass das Vorliegen einer PH bei Frühgeborenen stark mit der Sterblichkeit assoziiert ist (Risikoverhältnis 4,7), wobei die kumulative geschätzte Sterblichkeitsrate vor der Entlassung 16 % und in den ersten beiden Lebensjahren 40 % beträgt. Dieselben Metaanalysen ergaben jedoch, dass die meisten Berichte ausgewählte Patientenpopulationen untersuchten und es an genauen Schätzungen der Inzidenz- und Prävalenzraten im späteren Leben mangelt [
36
]. Es wurden noch keine Studien durchgeführt, um die Wirkung PH-spezifischer Therapien auf Kinder mit BPD offiziell zu bewerten.
Tabelle 4
bietet eine Zusammenfassung der entwicklungsbedingten Lungenerkrankungen, die das gemeinsame Merkmal entwicklungsbedingter Gefäßstörungen aufweisen.
TABELLE 4Entwicklungsstörungen der Lunge im Zusammenhang mit pulmonaler Hypertonie
Gruppe 4: PH aufgrund von LungenarterienobstruktionenChronische thromboembolische PH sind mit etwa 0–1,4 % der Fälle nach wie vor eine seltene Ursache für PAH bei Kindern [
22
,
26
]. Im Gegensatz dazu treten Lungenarterienobstruktionen bei einer Reihe von KHK auf, entweder angeboren oder erworben nach einer Korrekturoperation [
25
].Gruppe 5: PH mit unklaren und/oder multifaktoriellen MechanismenGruppe 5.4: Komplexe KHKDiese Gruppe umfasst hämatologische Störungen, systemische und metabolische Störungen und andere sowie komplexe KHK. Von besonderem Interesse für die pädiatrische Altersgruppe sind komplexe Herzerkrankungen, die mit angeborenen Anomalien des Lungengefäßsystems wie Segmentstörungen, Einzelventrikelphysiologie und dem Krummsäbelsyndrom einhergehen (
Tabelle 5
). PH ist in diesen Situationen äußerst schwer zu definieren oder zu klassifizieren [
37
]. Nach ausführlicher Diskussion auf der 5. WSPH im Jahr 2013 einigte sich die Pädiatrische Task Force darauf, mehrere Anomalien mit unterschiedlichem pulmonalem Blutfluss in die Kategorie „segmentale PH“ einzuordnen, was auf die besondere Natur dieser Entitäten im Vergleich zu anderen PH-Formen hinweist [
38
,
39
].
TABELLE 5Komplexe angeborene Herzfehler (Gruppe 5.4)
Während des aktuellen 6. WSPH erwog die Pädiatrische Task Force auch, Patienten mit der Physiologie eines einzelnen Ventrikels als eine weitere schwer zu definierende Gruppe einzubeziehen; Diese Gruppe nimmt weiter zu und stellt einen erheblichen Anteil der Patienten mit PVD in großen medizinischen Zentren dar. In verschiedenen Stadien können diese Patienten einen erhöhten oder verringerten pulmonalen Blutfluss aufweisen, und wenn sie ein Alter erreichen, das für das Fontan-Verfahren oder die vollständige kavo-pulmonale Verbindung geeignet ist, weisen diese Patienten unterschiedliche Grade von PVD und bronchopulmonalen Kollateralen auf. Anschließend induziert der chronische, nicht pulsierende Lungenkreislauf, der mit einem Fontan-Kreislauf einhergeht, wahrscheinlich eine sehr spezifische Form der PVD, die sich von der PVD bei anderen mit PH verbundenen Krankheiten unterscheidet [
40
]. Patienten mit Fontan-Kreislauf erfüllen in der Regel nicht die Definition einer PH mit mPAP >20–25 mmHg und wurden dementsprechend zuvor von der offiziellen WSPH-Klassifikation ausgeschlossen. Dennoch entwickeln diese Patienten eine PVD, die sich deutlich auf das Überleben auswirkt, und nach Angaben der Pediatric Task Force verdient die PVD im Rahmen der Physiologie einzelner Ventrikel eine Einbeziehung und wurde mit anderen Formen der PH der Gruppe 5 klassifiziert. Die Art und die Mechanismen, die der Pathobiologie der PVD in diesem Umfeld zugrunde liegen, bedürfen dringend weiterer Untersuchungen.In diesen komplexen KHK-Kategorien (Gruppe 5.4 ) reicht die allgemeine Definition von PH nicht aus und sollte angepasst werden. Derzeit gibt es keine ausreichenden Daten, die zeigen, dass zielgerichtete Therapien in dieser Population sicher und effizient sind, und weitere Studien sind erforderlich [
41
,
42
] .Zusätzliche besondere Überlegungen zur pädiatrischen klinischen KlassifizierungDie WSPH-Klassifikation für PH wurde ursprünglich für Erwachsene mit PH entwickelt. Der Grund für eine klinische Klassifizierung umfasst die Fähigkeit, die klinische Praxis zu stärken, einschließlich verbesserter Diagnose- und Managementstrategien, und dabei zu helfen, Leitlinien für die Priorisierung von Labor-, translationalen und epidemiologischen Forschungsfragen bereitzustellen. Darüber hinaus gehören zu den Zielen zur Verbesserung der Klassifizierungssysteme die Notwendigkeit der Klärung des Krankheitsphänotyps, die Förderung neuer Überlegungen zur Ursache und Krankheitspathobiologie, die Verbesserung diagnostischer Bewertungen, Verbesserungen der Korrelationen zwischen Phänotyp und therapeutischer Reaktionsfähigkeit sowie die Verbesserung des Designs klinischer Studien.Während sich gezeigt hat, dass die vom WSPH klassifizierten Hauptkategorien der PH auch bei Neugeborenen und Kindern hilfreich sind, gibt es anhaltende und wichtige Lücken, die berücksichtigt werden sollten, um ihren Nutzen in diesen spezifischen Altersgruppen zu verbessern.Im Jahr 2013 kam die erste WSPH Pediatric Task Force zu dem Schluss, dass eine gemeinsame Klassifizierung sowohl für Erwachsene als auch für Kinder bevorzugt wird, da Kinder mit PH, die im Neugeborenen- bis Jugendalter diagnostiziert wurden, nun bis ins Erwachsenenalter überleben und eine solche Klassifizierung den Übergang von der Pädiatrie zur Kinderkrankheit erleichtern wird Dienstleistungen für Erwachsene. Anschließend schlugen sie mehrere Änderungen vor, um Aspekte pädiatrischer Erkrankungen hervorzuheben und spezifische Merkmale der pädiatrischen PH im Kern der bestehenden Klassifikation besser zu berücksichtigen. Zu diesen Änderungen gehörten die Einstufung von PPHN als Unterklasse innerhalb von Störungen der Gruppe 1, eine detailliertere Kategorisierung von PAH bei KHK, die Hinzufügung des angeborenen Ein- und Ausflusstrakts des linken Herzens zu Gruppe 2 und die Einführung der Kategorie „entwicklungsbedingte Lungenerkrankung“. Gruppe 3 und von „segmentaler PH“ zu Gruppe 5.Im Jahr 2018 zielte die WSPH Pediatric Task Force darauf ab, spezifische pädiatrische Merkmale in der klinischen WSPH-Klassifikation weiter zu erfassen und gleichzeitig den Hauptkern der Klassifizierung, wie in Tabelle 2 des Task Force-Artikels von Simonneau et =inherital . dargelegt, beizubehalten . [
25
] in dieser Ausgabe des European Respiratory Journal . Sie schlugen zusätzliche, weitere Verfeinerungen dieser Gruppen vor, wie in den vorherigen Unterabschnitten besprochen, einschließlich einer separaten Bezeichnung für angeborene/erworbene Herz-Kreislauf-Erkrankungen, die zu postkapillärer PH führen (Gruppe 2.4), Entwicklungsstörungen der Lunge (Gruppe 3.5) und anderen Lungenarterienobstruktionen (Gruppe). 4.2) und komplexe KHK (Gruppe 5.4)Aktuelle pädiatrische Beobachtungsdaten zeigen, dass PAH bei „älteren“ Kindern trotz spezifischer Unterschiede viele Gemeinsamkeiten mit der PAH bei Erwachsenen aufweist. PH bei Neugeborenen ist jedoch häufig mit entwicklungsbedingten Gefäßanomalien und -reaktionen verbunden und wird in der aktuellen Klassifizierung der PH der Gruppe 3 zugeordnet, die mit Lungenerkrankungen und/oder Hypoxie einhergeht. Diese PVDs sind viel weniger mit PH bei Erwachsenen vergleichbar, da die Auswirkung von PH auf die unreife, sich entwickelnde Lunge als wesentlicher Faktor anerkannt wird, der für das Erscheinungsbild, die Diagnose, das Ansprechen auf die Therapie und das Ergebnis sowohl unmittelbar als auch langfristig von wesentlicher Bedeutung ist. Es ist klar, dass die PH bei Kindern und Erwachsenen zwar gemeinsame Merkmale aufweist, sich die Ätiologie, Epidemiologie und das Erscheinungsbild der PVD bei Neugeborenen und Kindern jedoch erheblich von denen bei Erwachsenen unterscheiden. Eines der charakteristischen Merkmale von PH bei Kindern ist die Schädigung des sich entwickelnden fetalen, neonatalen und pädiatrischen Lungenkreislaufs [
43
]. Ein weiteres Unterscheidungsmerkmal ist die häufige Assoziation mit Chromosomen-, genetischen und syndromalen Anomalien (11–52 %) sowie die phänotypischen Assoziationen, die in bis zu 33 % der Fälle zu multifaktoriellen Ursachen der PH führen können [
5
,
44
].Die aktuelle WSPH Pediatric Task Force schlug daher vor, die entwicklungsbedingten (vaskulären) Lungenerkrankungen als spezielle Unterkategorie innerhalb der Gruppe 3 PH (Gruppe 3.5) auszuweisen. Neben der anerkannten Häufigkeit und Bedeutung von PVD und PH bei Erkrankungen wie BPD und CDH umfasst diese Kategorie eine schnell wachsende Liste neu erkannter genetischer Entwicklungsstörungen der Lunge, darunter Surfactant-Anomalien, pulmonale interstitielle Glykogenese, alveoläre Kapillardysplasie, TBX4 -Mutationen usw andere (
Tabelle 4
).Pädiatrische PVDs gehen oft mit mehreren Komorbiditäten einher, die zum PH-Schweregrad beitragen und das Gesamtergebnis beeinflussen können. Wie bereits erwähnt, scheint sich der genetische Hintergrund der PH bei Kindern von dem der PH bei Erwachsenen zu unterscheiden, und begleitende genetische Störungen, Syndrome und Wachstumsstörungen kommen bei Kindern mit PH häufig vor. Ob diese letzteren als ursächlich zusammenhängend, als Krankheitsmodifikatoren oder als unschuldige Unbeteiligte anzusehen sind, ist oft nicht klar. Daher bleibt die genaue Phänotypisierung von Kindern mit PH und die Bewertung der Auswirkung von Komorbiditäten auf das Ergebnis bei jeder Klassifizierung von entscheidender Bedeutung.Down-SyndromEin anschauliches Beispiel für die komplexe Rolle von Komorbiditäten bei PH bei Kindern, das weitere Aufmerksamkeit erfordert, ist das Down-Syndrom. Diese pädiatrische Task Force diskutierte den einzigartigen klinischen Phänotyp von Neugeborenen, Säuglingen und Kindern mit Down-Syndrom und die mögliche Rolle des PH-Screenings in dieser Gruppe .Das Down-Syndrom oder Trisomie 21 ist bei Kindern mit einer erheblichen kardiovaskulären und pulmonalen Morbidität und Mortalität verbunden, einschließlich PH, chronischer Hypoxämie und wiederkehrenden Atemwegserkrankungen. Bei Neugeborenen mit Down-Syndrom besteht ein hohes Risiko, bei der Geburt eine schwere PPHN zu entwickeln, und es kommt oft zu einer aggressiveren PVD als Folge einer KHK oder einer Atemwegsobstruktion als bei Personen ohne Down-Syndrom. Die Mechanismen, die die Anfälligkeit von Säuglingen und Kindern mit Down-Syndrom für die Entwicklung eines schlechteren PH-Werts und einer Herz-Kreislauf-Erkrankung erhöhen, sind noch nicht vollständig verstanden. Frühere Studien haben gezeigt, dass Säuglinge, die mit dem Down-Syndrom sterben, Anzeichen einer Lungenhypoplasie aufweisen können, was sich in einer verminderten Alveolarisierung, peripheren Lungenzysten und einem Fortbestehen des Doppelkapillarnetzwerks zeigt [
45
]. Diese frühen Anomalien der gestoppten Lungenentwicklung können zu einer erhöhten Anfälligkeit für aggressivere Herz-Kreislauf- und Atemwegserkrankungen beim Down-Syndrom beitragen. Obwohl die Mechanismen für eine abnormale Lungen- und Lungengefäßentwicklung ungewiss sind, haben neuere Arbeiten gezeigt, dass drei antiangiogene Gene (antivaskulärer endothelialer Wachstumsfaktor) auf Chromosom 21 vorhanden sind und jeweils im menschlichen fetalen und kindlichen Lungengewebe überexprimiert werden, darunter Endostatin, RCAN -1 (Regulator von Calcineurin-1) und β-Amyloidpeptid. Experimentell gesehen verringert eine frühe Störung der angiogenen Signalübertragung das Gefäßwachstum und erhöht das Risiko für PH und beeinträchtigt auch das Wachstum des distalen Luftraums (Alveolar) [
46
,
47
]. Insgesamt deuten diese Labor- und klinischen Ergebnisse darauf hin, dass Personen mit Down-Syndrom sehr anfällig für ein vermindertes Lungengefäß- und Alveolarwachstum sind, was das Risiko für PH und Lungenhypoplasie erhöhen kann. Eine Beeinträchtigung des Lungengefäßwachstums kann das Risiko erhöhen, dass Umweltreize wie hämodynamischer Stress, insbesondere bei assoziierter KHK, intermittierende oder anhaltende Hypoxie mit obstruktiver Apnoe oder Lungenerkrankung, Virusinfektion, Aspiration und andere Faktoren, bei Patienten mit Down zu einer beschleunigten PH führen Syndrom als andere. Daher könnten die genetischen Folgen des Down-Syndroms mit einer Störung des Lungengefäß- und Alveolarwachstums verbunden sein, was darauf hindeutet, dass es sich beim Down-Syndrom um eine „entwicklungsbedingte Lungenerkrankung“ handelt. Die aktuelle WSPH Pediatric Task Force stimmte darin überein, dass der Phänotyp der Down-Syndrom-bedingten PH variabel ist und nicht allgemein in eine einzelne Klassifizierungsgruppe passt, dass Kinder mit Down-Syndrom jedoch in Gruppe 3 eingeteilt werden, wenn keine KHK vorliegt (Gruppe 1 bzw 2).Diagnose von PH bei KindernDa die Ätiologie der PH sehr vielfältig ist, ist ein methodischer und umfassender diagnostischer Ansatz von entscheidender Bedeutung, um einen genauen Diagnose- und Behandlungsplan zu erstellen. Darüber hinaus ist IPAH eine Diagnose „per Ausschluss“ und kann nur durch Ausschluss bekannter PH-Ursachen gestellt werden. Dennoch haben neuere Register gezeigt, dass die meisten Kinder keiner vollständigen Untersuchung unterzogen werden [
48
]. Ein aktualisierter umfassender pädiatrischer Diagnosealgorithmus ist in
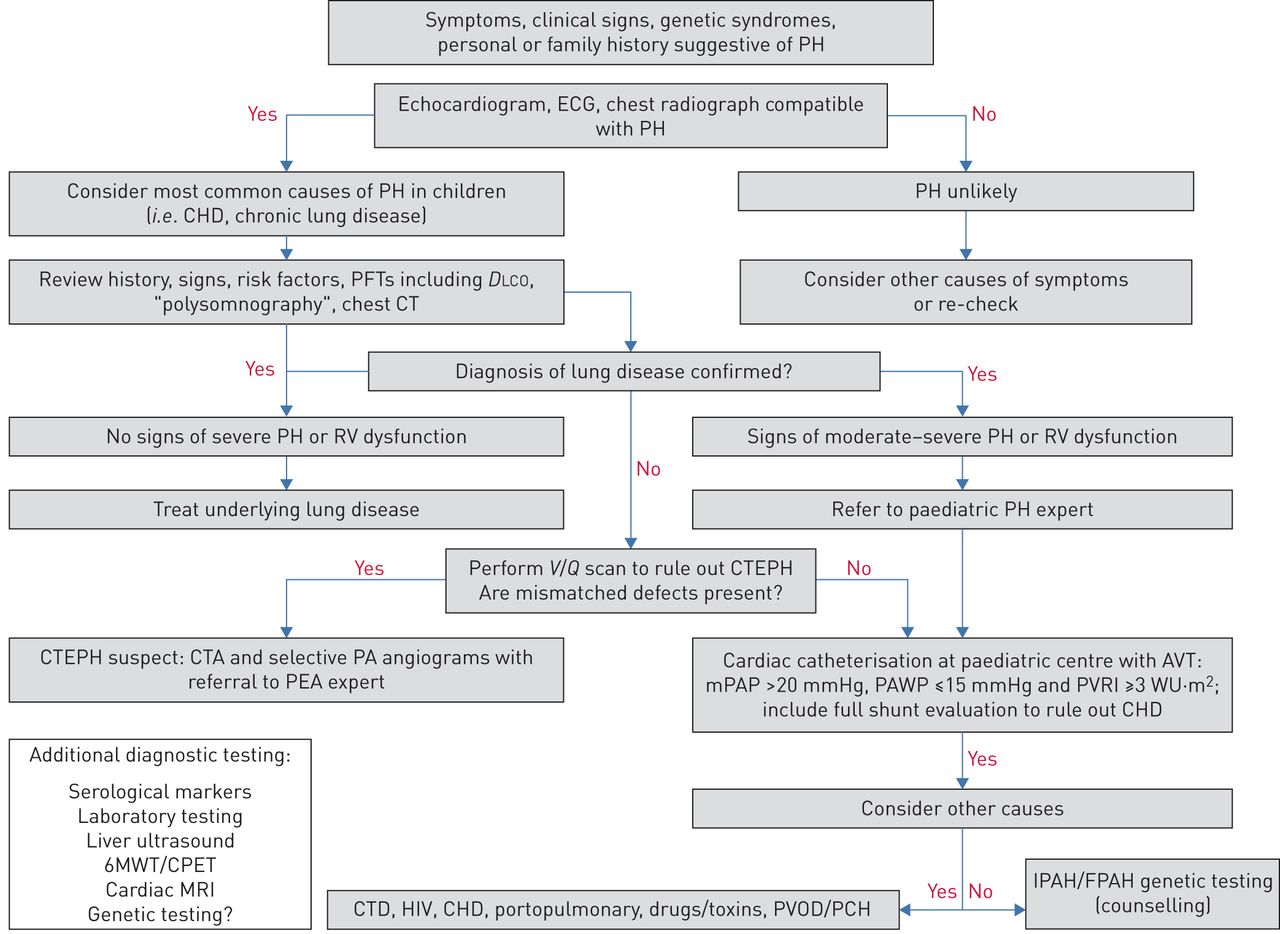
Abbildung 1
dargestellt . Besondere Situationen können die Entwicklung einer PAH begünstigen und sollten berücksichtigt werden.
=1emABBILDUNG 1Diagnosealgorithmus für pulmonale Hypertonie (PH) bei Kindern. KHK: angeborener Herzfehler; PFT: Lungenfunktionstest; D LCO : Diffusionskapazität der Lunge für Kohlenmonoxid; CT: Computertomographie; RV: rechtsventrikulär; V / Q : Ventilation/Perfusion; CTEPH: chronischer thromboembolischer PH; CTA: CT-Angiographie; PA: Lungenarterie; PEA: pulmonale Endarteriektomie; AVT: akuter Vasodilatatortest; mPAP: mittlerer pulmonalarterieller Druck; PAWP: pulmonaler arterieller Keildruck; PVRI: Lungengefäßwiderstandsindex; WU: Holzeinheiten; 6MWT: 6-minütiger Gehtest; CPET: kardiopulmonaler Belastungstest; MRT: Magnetresonanztomographie; CTD: Bindegewebserkrankung; PVOD: Lungenvenenverschlusskrankheit; PCH: pulmonale kapilläre Hämangiomatose; IPAH/FPAH: idiopathische/familiäre pulmonale arterielle Hypertonie. Katheterisierung des rechten HerzensDie Pädiatrie-Taskforce befasste sich mit mehreren Fragen zu den Risiken und Vorteilen der Rechtsherzkatheterisierung (RHC) zur Bestätigung der Diagnose von PH oder PAH bei Kindern und zu den Kindern, bei denen möglicherweise das höchste Risiko für unerwünschte Ereignisse während der RHC besteht.RHC bleibt der Goldstandard für die endgültige Diagnose und Art der PAH, die Durchführung einer AVT und die Bereitstellung nützlicher Daten für die Risikostratifizierung. Diese Notwendigkeit sollte mit den damit verbundenen Risiken abgewogen werden. Schwere Komplikationen im Zusammenhang mit RHC bei Kindern mit PH wurden mit 1–3 % angegeben und hängen im Allgemeinen mit dem klinischen Zustand und dem jungen Alter (Neugeborene und Kleinkinder) zusammen [
49–52
]
. Daher wird dringend empfohlen, die Herzkatheterisierung bei pädiatrischer PAH in erfahrenen pädiatrischen PH-Zentren durchzuführen, die Strategien zur Vermeidung dieser potenziellen Komplikationen anwenden und in der Lage sind, Komplikationen einschließlich PH-Krisen mit aggressiven Interventionen wie extrakorporaler Lebenserhaltung (ECLS) zu bewältigen. In seltenen Fällen kann es vorkommen, dass ein Kind zu krank ist, um sich sicher einer Herzkatheteruntersuchung zu unterziehen ( z. B. Funktionsklasse IV der Weltgesundheitsorganisation (WHO FC). In diesen Fällen, wenn der Verdacht auf PAH groß ist und die nicht-invasive Bildgebung sehr unterstützend ist, sollte man sich zunächst stabilisieren und unter sorgfältiger Beobachtung, meist auf der Intensivstation, vorsichtig eine geeignete PH-Therapie einleiten. RHC kann dann sicherer durchgeführt werden, wenn der Patient ausreichend stabilisiert ist. Es sollte jeder Versuch unternommen werden, Kinder mit IPAH/HPAH sicher einer RHC und AVT zu unterziehen, damit festgestellt werden kann, ob sie akut auf Vasoreaktivitätstests ansprechen und von einer CCB-Behandlung profitieren könnten. Für diejenigen, die robust reagieren, aber über eine schlechte Herzfunktion verfügen, würde ein CCB nicht verwendet werden, es sei denn, die Funktion verbessert sich.Die Indikationen für eine wiederholte Herzkatheterisierung bei Kindern mit PH sind nicht genau definiert, umfassen jedoch die Beurteilung des Behandlungseffekts, die klinische Verschlechterung, die Erkennung eines frühen Fortschreitens der Erkrankung, die Aufnahme in eine Lungentransplantation und die Vorhersage der Prognose. Es konnte jedoch nicht nachgewiesen werden, ob Änderungen der hämodynamischen Parameter mit einer Änderung des klinischen Ergebnisses einhergehen und ob diese Parameter daher nicht die Anforderungen erfüllen, um als etablierte Behandlungsziele zu dienen.Behandlungsstrategien und klinische EndpunkteDerzeit wird eine zielgerichtete Behandlungsstrategie für die Behandlung der pädiatrischen PAH vorgeschlagen. Trotz des Fehlens validierter Behandlungsziele bei pädiatrischer PAH wurden verschiedene Behandlungsrichtlinien für Kinder mit PAH vorgeschlagen, überwiegend basierend auf Expertenmeinungen
/url] 53–56 [url=https://erj.ersjournals.com/content/53/1/1801916#ref-53 . Ein offensichtlicher Mangel an Konsens in diesen Meinungen führte zu erheblichen Unterschieden in den gemeldeten pädiatrischen Behandlungsempfehlungen, was die Notwendigkeit von Beweisen unterstreicht.Im Jahr 2013 fasste die Pädiatrische Task Force der 5. WSPH die Determinanten eines höheren Risikos bei Kindern zusammen, darunter klinische Hinweise auf Rechtsherzversagen, Fortschreiten der Symptome, Synkope, Gedeihstörungen, WHO FC III oder IV, signifikant erhöhte oder steigende Natriuretika im Gehirn Peptid (BNP)-Spiegel, echokardiographische Anzeichen einer schweren Vergrößerung oder Dysfunktion des rechten Ventrikels, Perikarderguss und hämodynamische Parameter wie mPAP/mittlerer systemischer arterieller Druck (mSAP) > 0,75, mittlerer rechter Vorhofdruck (mRAP) > 10 mmHg und PVRI > 20 WU·m 2 [
54
]. Eine kürzlich durchgeführte systematische Überprüfung und Metaanalysen kamen zu dem Schluss, dass WHO FC, N-terminales Pro-BNP (NT-proBNP)/BNP, mRAP, PVRI, Herzindex und akute vasodilatatorische Reaktion durchweg als nützliche prognostische Faktoren für die Beurteilung langfristiger Ergebnisse angegeben werden bei pädiatrischer PAH und könnte daher zur anfänglichen Risikostratifizierung verwendet und als solche in Empfehlungen und Leitlinien aufgenommen werden [
57
,
58
].Allerdings sind Parameter mit prognostischer Aussagekraft nicht automatisch als Behandlungsziel geeignet. Behandlungsziele sind entweder klinisch bedeutsame Parameter, die widerspiegeln, wie sich ein Patient fühlt oder funktioniert, und können daher ein Ziel für die Behandlung sein, oder sie sollten Surrogate für das Überleben sein. Surrogate für das Überleben sind per Definition Parameter mit einer starken Korrelation zum Überleben, die durch die Behandlung verändert werden können, wobei eine solche Veränderung auf eine Verschlechterung oder Verbesserung der Krankheit hinweisen und eine Prognose für das langfristige Ergebnis darstellen sollte.WHO FC, das bei Kindern mit PAH angewendet wird und zeigt, wie sich das Kind fühlt und wie es funktioniert, hat sich nachweislich auch als starker Prädiktor für das transplantationsfreie Überleben erwiesen. Darüber hinaus wurde kürzlich gezeigt, dass die WHO-FC bei pädiatrischer PAH auch ein Surrogat für das Überleben ist, und als solche gilt die WHO-FC als Behandlungsziel, obwohl sie den Nachteil hat, dass es sich um eine potenziell subjektive Beurteilung handelt [
53
]. Ein speziell für Kinder konzipierter Funktionskurs wurde 2011 vorgeschlagen, wird jedoch aufgrund seiner Komplexität und des Einflusses durch Komorbiditäten immer noch in Betracht gezogen und hat daher noch keine breite Anwendung gefunden [
59
,
60
].Bei Erwachsenen mit PAH wurde der 6-Minuten-Gehtest (6MWT) zum Nachweis der Arzneimittelwirksamkeit eingesetzt. Obwohl seine Fähigkeit, als Ersatzendpunkt für späte Ergebnisse zu dienen, umstritten ist, kann der 6MWT als Behandlungsziel bei pädiatrischen Patienten nützlich sein, die in der Entwicklung sind und in der Lage sind, den Test durchzuführen (Kinder > 6 Jahre) [
61
,
62
]. Leider können jüngere Kinder den Test nicht zuverlässig durchführen, was dies zu einem suboptimalen primären Endpunkt in einer Studie über alle Altersgruppen von Kindern macht. Kardiopulmonale Belastungstests (CPET) stellen im Hinblick auf die Entwicklungsfähigkeiten sogar noch höhere Anforderungen und es fehlen pädiatrische Referenzwerte für CPET im Zusammenhang mit dem Ergebnis [
63
].Die Echokardiographie scheint ein offensichtliches Instrument zur Überwachung des Behandlungseffekts bei Kindern mit PH zu sein. Es ermöglicht eine funktionelle und strukturelle Beurteilung des Herzens sowie Schätzungen der pulmonalen Hämodynamik. Es ist weit verbreitet, nicht-invasiv und wird von Kindern gut vertragen. Leider unterliegt die Echokardiographie auch erheblichen Bediener- und Interpretationsschwankungen [
64
]. Während mehrere echokardiographische Variablen als Prädiktoren für das Ergebnis bei pädiatrischer PAH vorgeschlagen wurden, wurde heute gezeigt, dass nur die systolische Exkursion in der Trikuspidalringebene (TAPSE) einen starken Zusammenhang mit einem verbesserten Überleben während der Behandlung hat, was auf ihren potenziellen Nutzen als Behandlungsziel hinweist [
53
]. Neue echokardiographische Modalitäten zur Beurteilung der rechtsventrikulären Funktion (dreidimensionale Echokardiographie, Belastung und Belastungsrate sowie rechtsventrikuläre Schlagarbeit) sowie die Beurteilung des rechtsventrikulären Volumens und der Funktion durch Magnetresonanztomographie (MRT) sind hier vielversprechend, es liegen jedoch weitere Daten vor Die pädiatrische Bevölkerung wird benötigt, um den Wert dieser Techniken hinsichtlich der Festlegung eines prädiktiven Werts oder einer Leihmutterschaft für klinische Ergebnisse zu bestimmen [
65
,
66
].Zusätzlich zur rechtsventrikulären Funktion spiegelt die rechtsventrikuläre pulmonale Gefäßkopplung die rechtsventrikuläre Nachlast wider und gilt als wichtiges Maß für den kardiovaskulären Zustand und damit für die Prognose bei Patienten mit PH. MRT und Echokardiographie sind potenzielle Kandidaten für die nicht-invasive Überwachung dieses Kopplungszustands.Auf der pulmonalarteriellen Seite der ventrikulär-arteriellen Kopplung gewinnen Parameter der Lungenarteriensteifigkeit als prognostische Indikatoren bei PAH zunehmend an Interesse. Kürzlich wurde gezeigt, dass pulmonale Gefäßsteifheitsindizes die Entwicklung einer fortgeschrittenen PAH und die Mortalität bei pädiatrischer PVD vorhersagen können [
67–71
]
.Serumbiomarker haben den Vorteil, dass sie im peripheren Blut relativ leicht zu finden sind, und mehrere Biomarker wurden bei pädiatrischer PAH untersucht. Es wurde wiederholt gezeigt, dass zwei Serumbiomarker prognostische Fähigkeiten bei pädiatrischer PAH haben: NT-proBNP und Harnsäure. Eine aktuelle Metaanalyse bestätigte, dass NT-proBNP stark und konsistent mit dem Überleben von Kindern mit PAH korreliert und daher zur Risikostratifizierung in dieser Population verwendet werden kann. Um jedoch als Behandlungsziel oder klinischer Endpunkt verwendet zu werden, müssen Biomarker repräsentativ für den Krankheitsprozess und seine Entwicklung sein, was oft schwer nachzuweisen ist. Dennoch wurde kürzlich gezeigt, dass Veränderungen der NT-proBNP-Spiegel nach Behandlungsbeginn prädiktiv für das Überleben bei pädiatrischer PAH sind, was darauf hindeutet, dass NT-proBNP als Behandlungsziel in Frage kommt [
72
,
73
]. Zuvor wurde gezeigt, dass der Ausgangsharnsäurespiegel mit dem Überleben bei pädiatrischer PAH korreliert [
74
]. Kürzlich wurde gezeigt, dass die Entwicklung des Harnsäurespiegels im Laufe der Zeit mit dem Ergebnis bei pädiatrischer PAH korreliert [
75
]. Diese Ergebnisse zeigen, dass Harnsäure das Ergebnis nicht nur zu Studienbeginn, sondern auch während des Krankheitsverlaufs von PAH vorhersagen kann und daher möglicherweise auch als Behandlungsziel in Frage kommt.Kürzlich wurde die klinische Verschlechterung als zusammengesetzter Endpunkt für große randomisierte kontrollierte Studien (RCTs) bei Erwachsenen mit PAH eingeführt. Zu den Komponenten der klinischen Verschlechterung gehörten eindeutige Ereignisse wie Tod oder Lungentransplantation, die mit milderen Ereignissen wie Krankenhausaufenthalten, der Notwendigkeit einer zusätzlichen Therapie und einer Verschlechterung der Funktion einhergingen. Die Verwendung der klinischen Verschlechterung als Endpunkt wurde bei Erwachsenen mit PAH validiert, und es wurde gezeigt, dass die Endpunktkomponenten der leichten klinischen Verschlechterung eine hohe Vorhersagekraft für die spätere Mortalität haben. Diese Ergebnisse wurden nun in der pädiatrischen PAH-Population unter Verwendung klinischer Verschlechterungskomponenten reproduziert: Tod, Lungentransplantation, nicht elektive PAH-bedingte Krankenhauseinweisungen, einschließlich Krankenhauseinweisungen wegen Vorhofseptostomien, Beginn der intravenösen Prostanoidgabe und Funktionsverschlechterung (Verschlechterung der WHO-FC, ≥15). % Abnahme der 6MWD oder beides) [
76
]. Darüber hinaus zeigte diese Studie, dass bei pädiatrischer PAH eine klinische Verschlechterung mit einer hohen Ereignisrate auftrat, was darauf hindeutet, dass die klinische Verschlechterung als geeigneter Endpunkt in zukünftigen pädiatrischen Studien dienen könnte.Als alternatives Instrument zur Beurteilung der Funktionsfähigkeit von Kindern wurde auch die Überwachung der täglichen körperlichen Aktivität vorgeschlagen. Eine kürzlich durchgeführte Pilotstudie mit dreiachsiger Beschleunigungsmessung bei 29 Kindern mit PAH und 60 Kontrollpersonen zeigte, dass die körperliche Aktivität bei Kindern mit PAH deutlich verringert war und dass die Beschleunigungsmesser-Ausgabe mit klinischen Schweregradmarkern der Erkrankung und dem vorhergesagten Ergebnis korrelierte [
77
]. Größere Studien sind im Gange, um die Verwendung der Beschleunigungsmessung als klinisch bedeutsamen Endpunkt für klinische Studien zur pädiatrischen PAH zu validieren.Zusammenfassend lässt sich sagen, dass die neuen pädiatrischen Daten zunehmend Beweise und Unterstützung für das von der WSPH Pediatric Task Force im Jahr 2013 vorgeschlagene Risikostratifizierungsmodell liefern, mit einigen geringfügigen Änderungen im Jahr 2018. Beispielsweise konnte die prognostische Bedeutung von Synkopen nicht nachgewiesen werden und wird daher in Frage gestellt als Hochrisikofaktor für schlechte Ergebnisse. Wichtig ist, dass neue Erkenntnisse darauf hindeuten, dass bei pädiatrischer PAH das Streben nach einem Niedrigrisikoprofil unter Verwendung dieses WSPH-Tools zur pädiatrischen Risikobewertung auch als Behandlungsziel verwendet werden könnte, wie dies kürzlich bei Erwachsenen vorgeschlagen wurde
/url] 78–80 [url=https://erj.ersjournals.com/content/53/1/1801916#ref-78 .Aktualisierungen des pädiatrischen BehandlungsalgorithmusDie Prognose von Kindern mit PAH hat sich im letzten Jahrzehnt aufgrund neuer Therapeutika und aggressiver Behandlungsstrategien verbessert. Der Einsatz gezielter pulmonaler PAH-Therapien bei Kindern basiert jedoch fast ausschließlich auf Erfahrungen und Daten aus Studien an Erwachsenen und nicht auf Erkenntnissen aus klinischen Studien bei pädiatrischen Patienten. Aufgrund der komplexen Ätiologie und des relativen Mangels an Daten zu Kindern mit PAH bleibt die Auswahl geeigneter Therapien schwierig. Wir schlagen einen pragmatischen Behandlungsalgorithmus vor, der auf der Stärke der Expertenmeinung basiert und am besten auf Kinder mit IPAH anwendbar ist (
Abbildung 2
). Das ultimative Ziel der Behandlung sollte eine Verbesserung des Überlebens und die Erleichterung normaler Aktivitäten in der Kindheit ohne Selbstbeschränkung sein.
=1emFIGUR 2Behandlungsalgorithmus für pädiatrische idiopathische/familiäre pulmonale arterielle Hypertonie. CCB: Kalziumkanalblocker; ERA: Endothelin-Rezeptor-Agonist; PDE5i: Phosphodiesterase-Typ-5-Inhibitor. # : Verschlechterung oder Nichterreichen der Behandlungsziele. Eine Hintergrundtherapie mit Diuretika, Sauerstoff, Antikoagulation und Digoxin sollte individuell erwogen werden. Aufgrund der Vorlastabhängigkeit des rechten Ventrikels sollte darauf geachtet werden, das intravaskuläre Volumen nicht zu stark zu verringern. Nach der vollständigen Abklärung aller PH-Ursachen wird eine AVT empfohlen, um die Therapie festzulegen.
Bei Kindern mit
einer positiven AVT-Reaktion können orale CCBs eingeleitet werden [ 2–4
]
. Bei Kindern mit anhaltendem und verbessertem Ansprechen können CCBs fortgesetzt werden, aber der Zustand der Patienten kann sich verschlechtern, was eine wiederholte Untersuchung und zusätzliche Therapie erforderlich macht [
81
]. Klinische Erfahrungen legen nahe, dass diese Kinder zusätzlich zur gezielten PAH-Therapie weiterhin CCBs erhalten. Bei Kindern mit negativer AVT-Reaktion oder bei Kindern mit ausgefallener oder nicht anhaltender Reaktion auf CCBs sollte die Risikostratifizierung die zusätzliche Therapie bestimmen (
Tabelle 6
). Obwohl noch nicht bekannt ist, wie viele Kriterien mit geringerem oder höherem Risiko die therapeutische Wahl beeinflussen, sollte ein größerer Anteil von beiden als Rechtfertigung für eine Therapie angesehen werden. Wie bei erwachsenen Patienten gehören zu den Determinanten eines höheren Risikos bei Kindern klinische Anzeichen einer Rechtsherzinsuffizienz, ein Fortschreiten der Symptome, WHO FC III oder IV, signifikant erhöhte oder ansteigende BNP/NT-proBNP-Spiegel, eine schwere rechtsventrikuläre Vergrößerung oder Dysfunktion und ein Perikarderguss. Zu den weiteren hämodynamischen Parametern, die ein höheres Risiko vorhersagen, gehören das mPAP/mSAP-Verhältnis >0,75 [
82
], mRAP >10 mmHg und PVRI >20 WU·m 2 [
83
]. Weitere Hochrisikoparameter sind Gedeihstörungen. Bei Kindern mit negativer akuter Vasoreaktivitätsreaktion und geringerem Risiko wird der Beginn einer oralen Monotherapie empfohlen.
Die Behandlung der
Wahl ist ein Endothelin-Rezeptor-Antagonist (Bosentan [
83–90
], Ambrisentan [
91
,
92 ]) oder ein Phosphodiesterase-
Typ
-5-Hemmer (PDE5) (Sildenafil [ 93–100
]
, Tadalafil [
101
, 102
]
).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
TABELLE 6Determinanten des Risikos für idiopathische/erbliche pulmonale arterielle Hypertonie bei Kindern
Bei Kindern, deren Zustand sich unter der Behandlung mit Endothelin-Rezeptor-Antagonisten oder PDE5-Inhibitoren verschlechtert, kann es von Vorteil sein, eine frühe Kombinationstherapie (zusätzlich oder als Vorbehandlung) in Betracht zu ziehen. Bleibt das Kind in einer Niedrigrisikokategorie, kann die Zugabe von inhaliertem Prostazyklin (Iloprost [ [url=https://erj.ersjournals.com/content/ OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.