
- Beiträge: 1755
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Pulmonale Hypertonie: Die Lücke zwischen Erkennung und Behandlung schließen
Pulmonale Hypertonie: Die Lücke zwischen Erkennung und Behandlung schließen
08 Nov 2025 16:41 #2437
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Pulmonale Hypertonie: Die Lücke zwischen Erkennung und Behandlung schließen wurde erstellt von danny
www.acc.org/Latest-in-Cardiology/Article...y-HypertensionObwohl die pulmonale Hypertonie (PH) etwa 1 % der Bevölkerung betrifft und mit einem signifikanten Sterberisiko verbunden ist, wird sie im Vergleich zu anderen Herz-Kreislauf-Erkrankungen weiterhin zu selten diagnostiziert. Mehrere miteinander verbundene Faktoren tragen zu dieser klinischen Vernachlässigung bei.Doch mit einem neuen Medikament gegen pulmonale arterielle Hypertonie (PAH) und der zunehmenden Prävalenz von PH in Verbindung mit Linksherzerkrankungen (PH-LHD) aufgrund der Alterung der Bevölkerung ist es an der Zeit, der Erkennung von PH neuen Schwung zu verleihen und Kardiologen besser auf die Behandlung oder Mitbehandlung dieser Patienten vorzubereiten.Verzögerte oder verpasste PH-DiagnoseViele Patienten mit pulmonaler Hypertonie leiden monate- oder jahrelang unter fortschreitenden Symptomen, bevor eine korrekte Diagnose gestellt wird. In dieser Zeit kann die irreversible Umgestaltung der Lungengefäße unkontrolliert fortschreiten. Für diese Verzögerung gibt es mehrere Gründe.Zunächst einmal können Patienten mit pulmonaler Hypertonie (PH) unspezifische Symptome aufweisen, die fehldiagnostiziert oder ignoriert werden. Oftmals durchlaufen sie dann verschiedene Fachrichtungen, von der allgemeinen Kardiologie bis zur Pneumologie, bevor sie eine spezialisierte PH-Behandlung erhalten, was die Diagnose und Therapie weiter verzögert. „Da keine der beiden Fachrichtungen die alleinige Zuständigkeit für die Erkrankung hat, können Patienten durchs Raster fallen“, sagt Dr. Estefania Oliveros, Fachärztin für fortgeschrittene Herzinsuffizienz und Transplantationskardiologie (AHFTC) mit Schwerpunkt auf PH am Columbia University Irving Medical Center in New York.Zudem liegt der Schwerpunkt der kardiologischen Ausbildung traditionell und anhaltend auf linksseitigen Herzerkrankungen, und die meisten Ärzte fühlen sich bei der Interpretation der Hämodynamik des rechten Herzens unsicher. Darüber hinaus kann das komplexe Fünf-Gruppen-Klassifikationssystem der WHO ( Abbildung ) und die spezialisierten Behandlungsalgorithmen für Nicht-Spezialisten abschreckend wirken. Und das, bevor man die Komplexität der Diagnose und der Verordnung der Spezialmedikamente zur Behandlung der pulmonalen Hypertonie berücksichtigt, von denen 16 von der US-amerikanischen Arzneimittelbehörde (FDA) zugelassen sind.
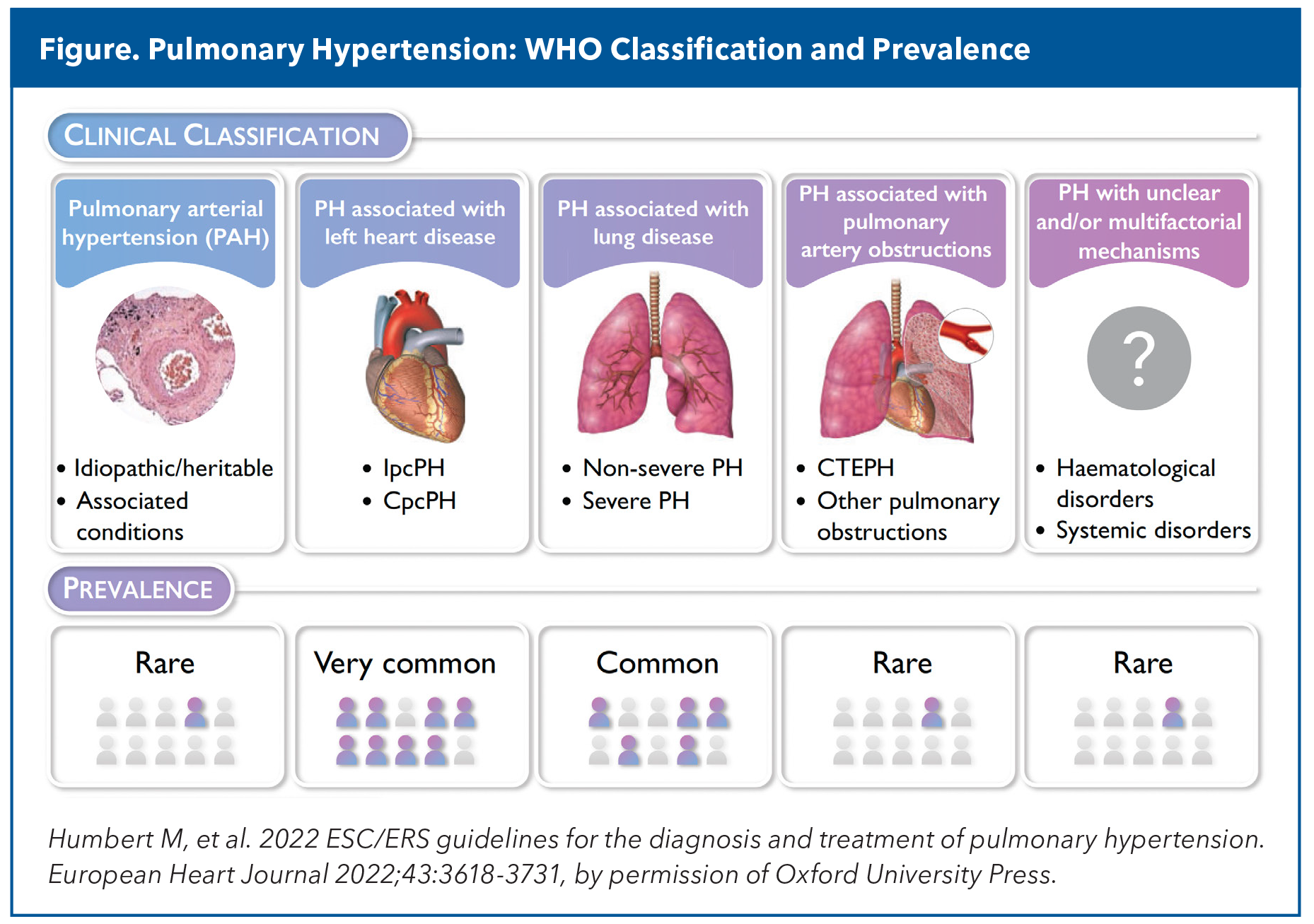 Angesichts all dessen ist es nicht ungewöhnlich, dass Patienten erst dann an einen Herzinsuffizienz-Spezialisten oder ein spezialisiertes Zentrum für pulmonale Hypertonie überwiesen werden, wenn sie bereits an Herzinsuffizienz leiden.PAH: Das Seltene, aber SichtbareDie pulmonale arterielle Hypertonie (PAH, auch Gruppe-1-PH genannt) ist eine seltene, aber fortschreitende Erkrankung. Sie betrifft 25 von 1 Million Menschen in westlichen Ländern, mit einer jährlichen Inzidenz von zwei bis fünf Fällen pro Million. PAH ist gekennzeichnet durch eine ausgeprägte Verengung und Versteifung des Lungengefäßbetts sowie einen fortschreitenden Anstieg der pulmonalen Gefäßbelastung, was zu einer Hypertrophie und einem Umbau des rechten Ventrikels führt.Trotz einiger Fortschritte und trotz mehr als einem Dutzend FDA-zugelassener Medikamente in verschiedenen Hauptkategorien (Endothelin-Rezeptorantagonisten, Stimulatoren der löslichen Guanylatzyklase, Phosphodiesterase-Hemmer, Prostacyclin-Analoga/Agonisten und die neueren Activin-Signalweg-Hemmer), die die körperliche Leistungsfähigkeit steigern, die Lebensqualität verbessern und das Fortschreiten der Krankheit verlangsamen, ist PAH immer noch mit einer signifikanten krankheitsbedingten Morbidität und Mortalität verbunden.Obwohl alle Formen der pulmonalen Hypertonie (PH) diagnostische Herausforderungen mit sich bringen, stellt die pulmonale arterielle Hypertonie (PAH) laut Oliveros die wohl größten Hürden dar. Im Gegensatz zur PH mit Linksherzinsuffizienz (PH-LHD), bei der eine zugrunde liegende Herzerkrankung einen klinischen Kontext für die Untersuchung bietet, gibt es bei der idiopathischen PAH keine offensichtliche Ursache, die weitere Untersuchungen erforderlich macht. Zudem tritt die Erkrankung häufig bei jüngeren Patienten auf – oft bei Frauen zwischen 30 und 60 Jahren, bei denen eine Herz-Kreislauf-Erkrankung in der Regel nicht vermutet wird. Dies kann insbesondere für Frauen im gebärfähigen Alter problematisch sein. Eine Schwangerschaft wird Patientinnen mit PAH generell nicht empfohlen, da sie mit einer hohen Sterblichkeitsrate von schätzungsweise 30–56 % einhergeht.
Angesichts all dessen ist es nicht ungewöhnlich, dass Patienten erst dann an einen Herzinsuffizienz-Spezialisten oder ein spezialisiertes Zentrum für pulmonale Hypertonie überwiesen werden, wenn sie bereits an Herzinsuffizienz leiden.PAH: Das Seltene, aber SichtbareDie pulmonale arterielle Hypertonie (PAH, auch Gruppe-1-PH genannt) ist eine seltene, aber fortschreitende Erkrankung. Sie betrifft 25 von 1 Million Menschen in westlichen Ländern, mit einer jährlichen Inzidenz von zwei bis fünf Fällen pro Million. PAH ist gekennzeichnet durch eine ausgeprägte Verengung und Versteifung des Lungengefäßbetts sowie einen fortschreitenden Anstieg der pulmonalen Gefäßbelastung, was zu einer Hypertrophie und einem Umbau des rechten Ventrikels führt.Trotz einiger Fortschritte und trotz mehr als einem Dutzend FDA-zugelassener Medikamente in verschiedenen Hauptkategorien (Endothelin-Rezeptorantagonisten, Stimulatoren der löslichen Guanylatzyklase, Phosphodiesterase-Hemmer, Prostacyclin-Analoga/Agonisten und die neueren Activin-Signalweg-Hemmer), die die körperliche Leistungsfähigkeit steigern, die Lebensqualität verbessern und das Fortschreiten der Krankheit verlangsamen, ist PAH immer noch mit einer signifikanten krankheitsbedingten Morbidität und Mortalität verbunden.Obwohl alle Formen der pulmonalen Hypertonie (PH) diagnostische Herausforderungen mit sich bringen, stellt die pulmonale arterielle Hypertonie (PAH) laut Oliveros die wohl größten Hürden dar. Im Gegensatz zur PH mit Linksherzinsuffizienz (PH-LHD), bei der eine zugrunde liegende Herzerkrankung einen klinischen Kontext für die Untersuchung bietet, gibt es bei der idiopathischen PAH keine offensichtliche Ursache, die weitere Untersuchungen erforderlich macht. Zudem tritt die Erkrankung häufig bei jüngeren Patienten auf – oft bei Frauen zwischen 30 und 60 Jahren, bei denen eine Herz-Kreislauf-Erkrankung in der Regel nicht vermutet wird. Dies kann insbesondere für Frauen im gebärfähigen Alter problematisch sein. Eine Schwangerschaft wird Patientinnen mit PAH generell nicht empfohlen, da sie mit einer hohen Sterblichkeitsrate von schätzungsweise 30–56 % einhergeht.
 „Das erste Problem ist, dass sie unter Atemnot und Schwellungen leiden, aber keine ärztliche Hilfe in Anspruch nehmen, weil sie relativ jung und ansonsten gesund sind“, sagt Oliveros. Das Durchschnittsalter von Patienten mit einer neu diagnostizierten pulmonalen arteriellen Hypertonie (PAH) in den aktuellen Registern beträgt 62 Jahre; Frauen sind weiterhin deutlich häufiger betroffen.„Das andere Problem ist, dass ihnen bei ihrem Hausarzt gesagt wird: ‚Oh, Sie haben Asthma, Allergien, körperliche Schwäche oder Angstzustände.‘ Bis sie dann einen Spezialisten für Herzinsuffizienz aufsuchen, befinden sie sich oft schon in der WHO-Funktionsklasse III oder IV.“Zu diesem Zeitpunkt ist die Prognose schlecht und die Behandlung, gelinde gesagt, belastend. „Je nach Schweregrad verabreichen wir entweder direkt intravenöse oder subkutane Infusionen oder eine Kombination aus Tabletten und Inhalatoren, die alle mit zahlreichen Nebenwirkungen verbunden sind“, erklärt Oliveros. „Und das alles noch bevor wir uns mit den Genehmigungen, der Spezialapotheke und all dem anderen auseinandersetzen müssen.“Der Sotatercept-DurchbruchDer bedeutendste Durchbruch in der PAH-Behandlung – möglicherweise überhaupt – gelang mit der FDA-Zulassung von Sotatercept im März 2024. Dieses Fusionsprotein und der erste Inhibitor der Activin-Signalübertragung stellen einen Paradigmenwechsel von der Symptombehandlung hin zur Krankheitsmodifikation dar.Im Gegensatz zu herkömmlichen PAH-Therapien, die primär die Vasokonstriktion mit sekundären Anti-Remodeling-Effekten behandeln, setzt Sotatercept an der grundlegenden Problematik der übermäßigen Zellproliferation in den Pulmonalarterien an, indem es das Gleichgewicht zwischen pro- und antiproliferativen Signalwegen verbessert. Präklinische Modelle zeigen zelluläre Veränderungen, die mit dünneren Gefäßwänden, einer teilweisen Rückbildung des rechtsventrikulären Remodelings und einer verbesserten Hämodynamik einhergehen.Dreifacher Beweis: Moderate, frühe und fortgeschrittene PAHDrei wegweisende klinische Phase-III-Studien haben die Sicherheit und Wirksamkeit von Sotatercept bei PAH belegt. Die erste, STELLAR, lieferte die grundlegenden Erkenntnisse, die zur FDA-Zulassung des Medikaments führten.<sup> 1 </sup> An dieser multizentrischen, doppelblinden Studie nahmen 323 Erwachsene mit PAH der WHO-Funktionsklasse II oder III teil, die eine stabile Basistherapie erhielten.Die subkutane Gabe von Sotatercept alle drei Wochen verbesserte die körperliche Leistungsfähigkeit im Vergleich zu Placebo signifikant (mediane Veränderung gegenüber dem Ausgangswert in Woche 24: 34,4 m vs. 1,0 m für Placebo; Differenz: 40,8 m; p<0,001). Gleichzeitig zeigten sich positive Effekte auf den pulmonalen Gefäßwiderstand, die WHO-Funktionsklasse und den NT-proBNP-Spiegel.Die zweite Phase-3-Studie mit dem Namen HYPERION untersuchte das Potenzial von Sotatercept bei neu diagnostizierten PAH-Patienten mit mittlerem oder hohem Risiko für eine Krankheitsprogression. An dieser globalen Studie nahmen 320 Patienten teil, bei denen die Diagnose innerhalb von 12 Monaten nach dem Screening gestellt wurde.Die HYPERION-Studie wurde vorzeitig abgebrochen, nachdem die in der STELLAR-Studie gezeigte überwältigende Wirksamkeit und die Veröffentlichung der Zwischenergebnisse der ZENITH-Studie zu einem Verlust der klinischen Äquivalenz geführt hatten. Die Studie erreichte ihren primären Endpunkt der Verkürzung der Zeit bis zur klinischen Verschlechterung; die vollständigen Ergebnisse werden zu einem späteren Zeitpunkt erwartet.Zum Zeitpunkt der Drucklegung von Cardiology wurden die vollständigen Ergebnisse der HYPERION-Studie im NEJM veröffentlicht.² Nach einer medianen Nachbeobachtungszeit von 13,2 Monaten zeigte sich eine klinische Verschlechterung bei 10,6 % der Patienten in der Sotatercept-Gruppe im Vergleich zu 36,9 % in der Placebo-Gruppe (Hazard Ratio 0,24; p < 0,001). Eine Verschlechterung der Belastbarkeit aufgrund von PAH wurde bei 5,0 % bzw. 28,8 % der Patienten beobachtet, ungeplante Krankenhausaufenthalte aufgrund einer PAH-Verschlechterung bei 1,9 % bzw. 8,8 %. Todesfälle jeglicher Ursache wurden bei 4,4 % der Patienten in der Sotatercept-Gruppe und bei 3,8 % in der Kontrollgruppe berichtet. Die häufigsten unerwünschten Ereignisse im Zusammenhang mit dem Studienmedikament waren Nasenbluten (31,9 %) und Teleangiektasien (26,2 %).Die ZENITH-Studie, die auf dem ACC.25-Kongress vorgestellt und gleichzeitig im NEJM veröffentlicht wurde , richtete sich an Hochrisikopatienten (WHO-Funktionsklasse III oder IV) mit hohem Sterberisiko innerhalb eines Jahres, die eine maximal tolerierte Basistherapie erhielten.<sup> 3 </sup> 172 Patienten erhielten alle drei Wochen zusätzlich Sotatercept oder Placebo. Der primäre Endpunkt der ZENITH-Studie umfasste Tod, Lungentransplantation oder PAH-bedingte Hospitalisierung.„ZENITH war wichtig, weil trotz der eindeutigen Erfolge früherer Studien mit Sotatercept Skepsis darüber bestand, ob Sotatercept oder irgendeine andere Therapie die Ergebnisse bei Patienten mit PAH im fortgeschrittenen Stadium, die oft als hochdifferenziert und klinisch instabil angesehen wird, sinnvoll verbessern könnte“, sagt Bradley A. Maron, MD, FACC , Direktor des Pulmonary Hypertension Program an der University of Maryland und ein führender Arzt-Wissenschaftler auf dem Gebiet der pulmonalen Hypertonie, dessen Arbeit dazu beigetragen hat, die Definition der pulmonalen Hypertonie zu verändern.Aufgrund der Ergebnisse der Zwischenanalyse des primären Endpunkts wurde die ZENITH-Studie wegen überwältigender Wirksamkeit vorzeitig beendet. Laut Merck wurde allen Teilnehmern, die die ZENITH-Studie abgeschlossen hatten, die Teilnahme an der offenen Verlängerungsstudie SOTERIA angeboten.Bei einer medianen Nachbeobachtungszeit von 10,6 Monaten reduzierte Sotatercept das relative Risiko schwerwiegender Morbiditäts- und Mortalitätsereignisse im Vergleich zu Placebo um 76 % (17,4 % vs. 54,7 %; Hazard Ratio 0,24; p < 0,001). Zum Zeitpunkt des Datenabschlusses traten Todesfälle jeglicher Ursache bei 8,1 % der Patienten in der Sotatercept-Gruppe und bei 15,1 % in der Placebo-Gruppe auf; Lungentransplantationen wurden bei 1,2 % bzw. 7,0 % der Patienten durchgeführt, und Krankenhausaufenthalte aufgrund einer Verschlechterung der pulmonalen arteriellen Hypertonie (PAH) erfolgten bei 9,3 % bzw. 50,0 % der Patienten.Aufgrund des konservativen Schwellenwerts für den p-Wert, der für den vorzeitigen Abbruch der Studie erforderlich war (p < 0,0021), war der Unterschied in der Sterblichkeitsrate nicht signifikant, wie Maron in einem Leitartikel anmerkte.⁴ „ Meiner Ansicht nach gab es ein klares Signal für einen Überlebensvorteil.“Die Behandlung ist mit einem erhöhten Risiko für Nasenbluten und Teleangiektasien verbunden, weshalb laut Maron weitere Überwachung erforderlich ist.
„Das erste Problem ist, dass sie unter Atemnot und Schwellungen leiden, aber keine ärztliche Hilfe in Anspruch nehmen, weil sie relativ jung und ansonsten gesund sind“, sagt Oliveros. Das Durchschnittsalter von Patienten mit einer neu diagnostizierten pulmonalen arteriellen Hypertonie (PAH) in den aktuellen Registern beträgt 62 Jahre; Frauen sind weiterhin deutlich häufiger betroffen.„Das andere Problem ist, dass ihnen bei ihrem Hausarzt gesagt wird: ‚Oh, Sie haben Asthma, Allergien, körperliche Schwäche oder Angstzustände.‘ Bis sie dann einen Spezialisten für Herzinsuffizienz aufsuchen, befinden sie sich oft schon in der WHO-Funktionsklasse III oder IV.“Zu diesem Zeitpunkt ist die Prognose schlecht und die Behandlung, gelinde gesagt, belastend. „Je nach Schweregrad verabreichen wir entweder direkt intravenöse oder subkutane Infusionen oder eine Kombination aus Tabletten und Inhalatoren, die alle mit zahlreichen Nebenwirkungen verbunden sind“, erklärt Oliveros. „Und das alles noch bevor wir uns mit den Genehmigungen, der Spezialapotheke und all dem anderen auseinandersetzen müssen.“Der Sotatercept-DurchbruchDer bedeutendste Durchbruch in der PAH-Behandlung – möglicherweise überhaupt – gelang mit der FDA-Zulassung von Sotatercept im März 2024. Dieses Fusionsprotein und der erste Inhibitor der Activin-Signalübertragung stellen einen Paradigmenwechsel von der Symptombehandlung hin zur Krankheitsmodifikation dar.Im Gegensatz zu herkömmlichen PAH-Therapien, die primär die Vasokonstriktion mit sekundären Anti-Remodeling-Effekten behandeln, setzt Sotatercept an der grundlegenden Problematik der übermäßigen Zellproliferation in den Pulmonalarterien an, indem es das Gleichgewicht zwischen pro- und antiproliferativen Signalwegen verbessert. Präklinische Modelle zeigen zelluläre Veränderungen, die mit dünneren Gefäßwänden, einer teilweisen Rückbildung des rechtsventrikulären Remodelings und einer verbesserten Hämodynamik einhergehen.Dreifacher Beweis: Moderate, frühe und fortgeschrittene PAHDrei wegweisende klinische Phase-III-Studien haben die Sicherheit und Wirksamkeit von Sotatercept bei PAH belegt. Die erste, STELLAR, lieferte die grundlegenden Erkenntnisse, die zur FDA-Zulassung des Medikaments führten.<sup> 1 </sup> An dieser multizentrischen, doppelblinden Studie nahmen 323 Erwachsene mit PAH der WHO-Funktionsklasse II oder III teil, die eine stabile Basistherapie erhielten.Die subkutane Gabe von Sotatercept alle drei Wochen verbesserte die körperliche Leistungsfähigkeit im Vergleich zu Placebo signifikant (mediane Veränderung gegenüber dem Ausgangswert in Woche 24: 34,4 m vs. 1,0 m für Placebo; Differenz: 40,8 m; p<0,001). Gleichzeitig zeigten sich positive Effekte auf den pulmonalen Gefäßwiderstand, die WHO-Funktionsklasse und den NT-proBNP-Spiegel.Die zweite Phase-3-Studie mit dem Namen HYPERION untersuchte das Potenzial von Sotatercept bei neu diagnostizierten PAH-Patienten mit mittlerem oder hohem Risiko für eine Krankheitsprogression. An dieser globalen Studie nahmen 320 Patienten teil, bei denen die Diagnose innerhalb von 12 Monaten nach dem Screening gestellt wurde.Die HYPERION-Studie wurde vorzeitig abgebrochen, nachdem die in der STELLAR-Studie gezeigte überwältigende Wirksamkeit und die Veröffentlichung der Zwischenergebnisse der ZENITH-Studie zu einem Verlust der klinischen Äquivalenz geführt hatten. Die Studie erreichte ihren primären Endpunkt der Verkürzung der Zeit bis zur klinischen Verschlechterung; die vollständigen Ergebnisse werden zu einem späteren Zeitpunkt erwartet.Zum Zeitpunkt der Drucklegung von Cardiology wurden die vollständigen Ergebnisse der HYPERION-Studie im NEJM veröffentlicht.² Nach einer medianen Nachbeobachtungszeit von 13,2 Monaten zeigte sich eine klinische Verschlechterung bei 10,6 % der Patienten in der Sotatercept-Gruppe im Vergleich zu 36,9 % in der Placebo-Gruppe (Hazard Ratio 0,24; p < 0,001). Eine Verschlechterung der Belastbarkeit aufgrund von PAH wurde bei 5,0 % bzw. 28,8 % der Patienten beobachtet, ungeplante Krankenhausaufenthalte aufgrund einer PAH-Verschlechterung bei 1,9 % bzw. 8,8 %. Todesfälle jeglicher Ursache wurden bei 4,4 % der Patienten in der Sotatercept-Gruppe und bei 3,8 % in der Kontrollgruppe berichtet. Die häufigsten unerwünschten Ereignisse im Zusammenhang mit dem Studienmedikament waren Nasenbluten (31,9 %) und Teleangiektasien (26,2 %).Die ZENITH-Studie, die auf dem ACC.25-Kongress vorgestellt und gleichzeitig im NEJM veröffentlicht wurde , richtete sich an Hochrisikopatienten (WHO-Funktionsklasse III oder IV) mit hohem Sterberisiko innerhalb eines Jahres, die eine maximal tolerierte Basistherapie erhielten.<sup> 3 </sup> 172 Patienten erhielten alle drei Wochen zusätzlich Sotatercept oder Placebo. Der primäre Endpunkt der ZENITH-Studie umfasste Tod, Lungentransplantation oder PAH-bedingte Hospitalisierung.„ZENITH war wichtig, weil trotz der eindeutigen Erfolge früherer Studien mit Sotatercept Skepsis darüber bestand, ob Sotatercept oder irgendeine andere Therapie die Ergebnisse bei Patienten mit PAH im fortgeschrittenen Stadium, die oft als hochdifferenziert und klinisch instabil angesehen wird, sinnvoll verbessern könnte“, sagt Bradley A. Maron, MD, FACC , Direktor des Pulmonary Hypertension Program an der University of Maryland und ein führender Arzt-Wissenschaftler auf dem Gebiet der pulmonalen Hypertonie, dessen Arbeit dazu beigetragen hat, die Definition der pulmonalen Hypertonie zu verändern.Aufgrund der Ergebnisse der Zwischenanalyse des primären Endpunkts wurde die ZENITH-Studie wegen überwältigender Wirksamkeit vorzeitig beendet. Laut Merck wurde allen Teilnehmern, die die ZENITH-Studie abgeschlossen hatten, die Teilnahme an der offenen Verlängerungsstudie SOTERIA angeboten.Bei einer medianen Nachbeobachtungszeit von 10,6 Monaten reduzierte Sotatercept das relative Risiko schwerwiegender Morbiditäts- und Mortalitätsereignisse im Vergleich zu Placebo um 76 % (17,4 % vs. 54,7 %; Hazard Ratio 0,24; p < 0,001). Zum Zeitpunkt des Datenabschlusses traten Todesfälle jeglicher Ursache bei 8,1 % der Patienten in der Sotatercept-Gruppe und bei 15,1 % in der Placebo-Gruppe auf; Lungentransplantationen wurden bei 1,2 % bzw. 7,0 % der Patienten durchgeführt, und Krankenhausaufenthalte aufgrund einer Verschlechterung der pulmonalen arteriellen Hypertonie (PAH) erfolgten bei 9,3 % bzw. 50,0 % der Patienten.Aufgrund des konservativen Schwellenwerts für den p-Wert, der für den vorzeitigen Abbruch der Studie erforderlich war (p < 0,0021), war der Unterschied in der Sterblichkeitsrate nicht signifikant, wie Maron in einem Leitartikel anmerkte.⁴ „ Meiner Ansicht nach gab es ein klares Signal für einen Überlebensvorteil.“Die Behandlung ist mit einem erhöhten Risiko für Nasenbluten und Teleangiektasien verbunden, weshalb laut Maron weitere Überwachung erforderlich ist.
 „Sotatercept ist eindeutig ein großer Fortschritt und hat, zusammen mit anderen Innovationen und Weiterentwicklungen auf diesem Gebiet, PAH wirklich aus der Dunkelheit geholt – von einer Krankheit, die unbehandelbar und stets tödlich war, zu einer Krankheit, für die es eine Vielzahl verschiedener Optionen gibt“, sagt Maron.„Deshalb müssen Kardiologen sich dieser Krankheit stärker bewusst sein, eine geringere Verdachtswahrscheinlichkeit für ihre Diagnose haben und darauf vorbereitet sein, Patienten zu sehen, die diese Medikamente einnehmen“, fügt er hinzu.Taten sprechen lassenOliveros hat die Vorteile von Sotatercept aus nächster Nähe miterlebt. Eine Patientin, die zuvor auf eine Infusionspumpe und zwei orale Medikamente angewiesen war und nach einem Termin nicht einmal mehr vom Büro zu ihrem Auto laufen konnte, konnte innerhalb von vier Monaten nach Beginn der Sotatercept-Therapie wieder im Garten arbeiten.CTEPH: Die "heilbare" Form der PHDie chronische thromboembolische pulmonale Hypertonie (CTEPH), auch Gruppe 4 der pulmonalen Hypertonie gemäß der revidierten klinischen Klassifikation genannt, entsteht, wenn chronische Blutgerinnsel die Lungenarterien verstopfen und Narbengewebe bilden, das den Blutfluss beeinträchtigt. Im Gegensatz zu akuten Lungenembolien (LE), die durch Antikoagulation aufgelöst werden können, erfordern diese organisierten Thromben eine operative Entfernung.Unter den fünf WHO-Gruppen der pulmonalen Hypertonie (PH) ist die chronisch-thromboembolische pulmonale Hypertonie (CTEPH) die einzige potenziell heilbare Form. Diese Besonderheit hat in der Post-COVID-Ära an klinischer Bedeutung gewonnen, da die thrombotischen Komplikationen der Pandemie wichtige Fragen zu den langfristigen kardiovaskulären Folgen aufwerfen.Diese Erkrankung betrifft schätzungsweise 3 % der Patienten nach einer akuten Lungenembolie (5 % bei Asiaten), wobei Experten davon ausgehen, dass sie weiterhin deutlich unterdiagnostiziert ist.⁶„Wenn ein Patient im 6-Minuten-Gehtest 60 Meter mehr schafft, kann er beispielsweise seinen Wocheneinkauf ohne ständige Pausen erledigen. Das ist für ihn wirklich ein großer Vorteil!“, sagt Oliveros. „Das Nebenwirkungsprofil von Sotatercept ist sehr gut. Die schlimmsten Nebenwirkungen sind Kopfschmerzen oder Teleangiektasien – die zwar lästig sein können –, aber insgesamt gesehen ist das natürlich nicht so schlimm.“Die hohen Kosten der PAH-Therapien, insbesondere neuer Wirkstoffe wie Sotatercept, werfen wichtige Fragen hinsichtlich des Patientenzugangs und der Chancengleichheit im Gesundheitswesen auf. Sotatercept kostet derzeit zwischen 25.000 und 30.000 US-Dollar pro Monat und muss lebenslang eingenommen werden; aktuell liegen keine Daten zu den Folgen nach Absetzen des Medikaments vor.Die schweigende Mehrheit: PH-LHDWährend Sotatercept derzeit die Schlagzeilen dominiert, dürfen Kardiologen das eigentliche Problem nicht aus den Augen verlieren: PH-LHD, die mit Abstand häufigste Form der Erkrankung in der klinischen Praxis.„Wenn man sich PH als Kuchen vorstellt, macht PH-LHD 65–80 % des Kuchens aus, während PAH nur etwa 10 % ausmacht“, sagt Dr. Onyedika J. Ilonze, FACC . Er ist Spezialist für Herzinsuffizienz und Transplantation an der Indiana University, IU Health Methodist Professional Center, und Mitglied der Sektion Herzinsuffizienz und Transplantation des American College of Cardiac Cancer (ACC). Die pulmonale Hypertonie bei Linksherzinsuffizienz (PH-LHD) ist mit einer hohen Mortalität verbunden, wird aber häufig unterdiagnostiziert. Die heterogene Patientengruppe umfasst Patienten mit Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF), Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) und Herzklappenerkrankungen. Die Erkrankung wird weiter in isolierte postkapilläre PH (Ipc-PH) und kombinierte post- und präkapilläre PH (Cpc-PH) unterteilt.Ein Zusammenhang mit COVID-19, den es wert ist, beobachtet zu werden
„Sotatercept ist eindeutig ein großer Fortschritt und hat, zusammen mit anderen Innovationen und Weiterentwicklungen auf diesem Gebiet, PAH wirklich aus der Dunkelheit geholt – von einer Krankheit, die unbehandelbar und stets tödlich war, zu einer Krankheit, für die es eine Vielzahl verschiedener Optionen gibt“, sagt Maron.„Deshalb müssen Kardiologen sich dieser Krankheit stärker bewusst sein, eine geringere Verdachtswahrscheinlichkeit für ihre Diagnose haben und darauf vorbereitet sein, Patienten zu sehen, die diese Medikamente einnehmen“, fügt er hinzu.Taten sprechen lassenOliveros hat die Vorteile von Sotatercept aus nächster Nähe miterlebt. Eine Patientin, die zuvor auf eine Infusionspumpe und zwei orale Medikamente angewiesen war und nach einem Termin nicht einmal mehr vom Büro zu ihrem Auto laufen konnte, konnte innerhalb von vier Monaten nach Beginn der Sotatercept-Therapie wieder im Garten arbeiten.CTEPH: Die "heilbare" Form der PHDie chronische thromboembolische pulmonale Hypertonie (CTEPH), auch Gruppe 4 der pulmonalen Hypertonie gemäß der revidierten klinischen Klassifikation genannt, entsteht, wenn chronische Blutgerinnsel die Lungenarterien verstopfen und Narbengewebe bilden, das den Blutfluss beeinträchtigt. Im Gegensatz zu akuten Lungenembolien (LE), die durch Antikoagulation aufgelöst werden können, erfordern diese organisierten Thromben eine operative Entfernung.Unter den fünf WHO-Gruppen der pulmonalen Hypertonie (PH) ist die chronisch-thromboembolische pulmonale Hypertonie (CTEPH) die einzige potenziell heilbare Form. Diese Besonderheit hat in der Post-COVID-Ära an klinischer Bedeutung gewonnen, da die thrombotischen Komplikationen der Pandemie wichtige Fragen zu den langfristigen kardiovaskulären Folgen aufwerfen.Diese Erkrankung betrifft schätzungsweise 3 % der Patienten nach einer akuten Lungenembolie (5 % bei Asiaten), wobei Experten davon ausgehen, dass sie weiterhin deutlich unterdiagnostiziert ist.⁶„Wenn ein Patient im 6-Minuten-Gehtest 60 Meter mehr schafft, kann er beispielsweise seinen Wocheneinkauf ohne ständige Pausen erledigen. Das ist für ihn wirklich ein großer Vorteil!“, sagt Oliveros. „Das Nebenwirkungsprofil von Sotatercept ist sehr gut. Die schlimmsten Nebenwirkungen sind Kopfschmerzen oder Teleangiektasien – die zwar lästig sein können –, aber insgesamt gesehen ist das natürlich nicht so schlimm.“Die hohen Kosten der PAH-Therapien, insbesondere neuer Wirkstoffe wie Sotatercept, werfen wichtige Fragen hinsichtlich des Patientenzugangs und der Chancengleichheit im Gesundheitswesen auf. Sotatercept kostet derzeit zwischen 25.000 und 30.000 US-Dollar pro Monat und muss lebenslang eingenommen werden; aktuell liegen keine Daten zu den Folgen nach Absetzen des Medikaments vor.Die schweigende Mehrheit: PH-LHDWährend Sotatercept derzeit die Schlagzeilen dominiert, dürfen Kardiologen das eigentliche Problem nicht aus den Augen verlieren: PH-LHD, die mit Abstand häufigste Form der Erkrankung in der klinischen Praxis.„Wenn man sich PH als Kuchen vorstellt, macht PH-LHD 65–80 % des Kuchens aus, während PAH nur etwa 10 % ausmacht“, sagt Dr. Onyedika J. Ilonze, FACC . Er ist Spezialist für Herzinsuffizienz und Transplantation an der Indiana University, IU Health Methodist Professional Center, und Mitglied der Sektion Herzinsuffizienz und Transplantation des American College of Cardiac Cancer (ACC). Die pulmonale Hypertonie bei Linksherzinsuffizienz (PH-LHD) ist mit einer hohen Mortalität verbunden, wird aber häufig unterdiagnostiziert. Die heterogene Patientengruppe umfasst Patienten mit Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF), Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) und Herzklappenerkrankungen. Die Erkrankung wird weiter in isolierte postkapilläre PH (Ipc-PH) und kombinierte post- und präkapilläre PH (Cpc-PH) unterteilt.Ein Zusammenhang mit COVID-19, den es wert ist, beobachtet zu werden
 Der gut belegte Zusammenhang zwischen COVID-19 und Hyperkoagulabilität sowie Lungenembolie hat Bedenken hinsichtlich eines möglichen Anstiegs der CTEPH-Inzidenz geweckt.<sup> 7,8</sup> Das Virus löst einen prothrombotischen Zustand aus, der über die akute Erkrankung hinaus anhalten kann; dokumentierte Fälle von Lungenembolie traten Wochen bis Monate nach der Erstinfektion auf.Obwohl die endgültigen epidemiologischen Daten zur Inzidenz von CTEPH nach COVID-19 noch ausstehen, ist der zeitliche Zusammenhang besorgniserregend. Da sich CTEPH typischerweise sechs Monate bis zwei Jahre nach einem initialen embolischen Ereignis entwickelt, stehen wir möglicherweise erst am Anfang der vollen kardiovaskulären Folgen der Höhepunkte der Pandemie.„Wir wissen es noch nicht genau, aber wir werden dies möglicherweise häufiger beobachten, wenn die Menschen einige Jahre länger von COVID-19 verschont bleiben. Wir gehen aber auch davon aus, dass CTEPH generell unterschätzt wird und dass es viele Menschen gibt, die möglicherweise an einer schlechten Atemwegsgesundheit leiden, dieses Narbengewebe haben und es einfach nicht wissen“, sagt Oliveros.Das Besondere an der chronisch-thromboembolischen pulmonalen Hypertonie (CTEPH) unter den PH-Subtypen ist die Möglichkeit der chirurgischen Heilung durch pulmonale Thromboendarterektomie (PTE). Bei diesem komplexen Eingriff, der in spezialisierten Zentren durchgeführt wird, werden organisierte Blutgerinnsel unter tiefer Hypothermie und Kreislaufstillstand entfernt.Die jüngsten Ergebnisse der PTE sind vielversprechend: Eine Metaanalyse aus dem Jahr 2025 berichtet von einer Mortalität von 8,4 % über alle Zentren hinweg, die in spezialisierten Zentren jedoch auf 1–2 % sinkt.⁶ UC San Diego Health, wo das Verfahren entwickelt wurde, hat bereits über 5.000 PTEs mit hervorragenden Ergebnissen durchgeführt.„Eine meiner Sorgen ist, dass einige Zentren, die nicht ausreichend auf die Behandlung dieser Erkrankung vorbereitet sind, versuchen könnten, das Blutgerinnsel zu entfernen, weil sie es für ein akutes, frisches Gerinnsel halten, es aber nicht vollständig entfernen. Sie entfernen möglicherweise nur einen Teil davon und verschließen die Wunde wieder“, bemerkt Oliveros. „Daher besteht bei dieser speziellen Patientengruppe Bedarf an multidisziplinär ausgebildeten Teams, bestehend aus einem Spezialisten für pulmonale Hypertonie, einem Radiologen, einem Chirurgen und einem interventionellen Kardiologen, die alle in der Diagnose und Behandlung dieser spezifischen Erkrankung geschult sind.“Für Patienten, die für eine PTE nicht geeignet sind, stellen die Ballon-Pulmonalangioplastie und die gezielte medikamentöse Therapie mit Riociguat sinnvolle Alternativen dar.Die COVID-19-Pandemie unterstreicht die Bedeutung der Aufklärung über CTEPH. Die Herausforderung besteht in der Erkennung: Viele COVID-bedingte embolische Ereignisse verlaufen möglicherweise subklinisch, und der schleichende Beginn von CTEPH-Symptomen – fortschreitende Atemnot, Müdigkeit und Belastungsintoleranz – kann leicht fälschlicherweise als „Long COVID“ oder Dekonditionierung interpretiert werden.„Manche Menschen haben ausgedehnte chronische Blutgerinnsel in der Lunge, ohne es zu wissen, sind aber nicht in der Lage, einen Raum zu durchqueren. Wir führen eine PTE durch und entfernen das gesamte Gerinnsel, woraufhin sich ihr Zustand dramatisch verbessert“, sagt Oliveros.„Da die Menschen immer älter werden, steigt auch die Wahrscheinlichkeit für Erkrankungen des linken Herzens, sei es durch Kardiomyopathie, Herzinsuffizienz oder Herzklappenerkrankungen, was wiederum das Risiko für pulmonale Hypertonie erhöht“, sagt Ilonze, Erstautor eines kürzlich in JACC: Heart Failure veröffentlichten Übersichtsartikels zum Thema pulmonale Hypertonie bei Linksherzerkrankungen.⁵Er vermutet, dass Ärzte nicht ausreichend darüber nachdenken, ob ihre Patienten mit Linksherzpathologie möglicherweise auch an pulmonaler Hypertonie (PH) leiden. Symptome der PH in Verbindung mit einer Linksherzerkrankung (Atemnot, Müdigkeit, Belastungsintoleranz) werden oft ausschließlich der zugrunde liegenden Linksherzerkrankung zugeschrieben, wobei Ärzte annehmen, dass sich verschlimmernde Symptome eher auf das Fortschreiten der Herzinsuffizienz als auf die Entwicklung einer begleitenden PH in Verbindung mit einer Linksherzerkrankung zurückzuführen sind.„Ein neues Medikament gegen PAH zu haben, ist aufregend, aber es verschärft das Problem wahrscheinlich noch etwas. Es besteht immer die Tendenz, nach der Krankheit zu suchen, für die es ein vielversprechendes neues Medikament gibt, und nicht nach der Krankheit, für die man dem Patienten nichts Besonderes anbieten kann.“Ein weiteres mögliches Hindernis für eine korrekte Diagnose ist der therapeutische Nihilismus: Da pulmonale Vasodilatatoren bei PH-LHD kontraindiziert sind und das neu zugelassene und äußerst wirksame Sotatercept nur für PAH zugelassen ist, könnten einige Kliniker der Ansicht sein, dass der therapeutische Nutzen einer spezifischen Diagnose begrenzt ist, was zu einer weniger aggressiven Diagnostik führt.Wenn bei Patienten bereits eine Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF), Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) oder eine Herzklappenerkrankung diagnostiziert wurde, besteht weniger Anreiz für eine invasive hämodynamische Untersuchung. „Die Denkweise ändert sich: ‚Wir wissen ja bereits, dass sie eine Herzerkrankung haben‘, anstatt die pulmonale Gefäßkomponente zu untersuchen“, erklärt Ilonze. Dies führt dazu, dass die pulmonale Hypertonie in Kombination mit einer Linksherzerkrankung (LHD) unerkannt und unbehandelt bleibt. „Es ist wichtig zu erkennen, dass die Entwicklung einer pulmonalen Hypertonie zusätzlich zur LHD die Prognose der bereits bestehenden LHD verschlechtert“, fügt er hinzu.Maron weist darauf hin, dass es zwei Patientengruppen gibt, bei denen eine „strenge Vorgehensweise bei pulmonaler Hypertonie“ besonders wichtig ist. Die erste Gruppe umfasst Patienten mit Mitralklappenerkrankung, bei denen das Vorliegen einer pulmonalen Hypertonie aufgrund der Erkrankung eine Indikation für eine Operation darstellen kann, und die zweite Gruppe betrifft Patienten, die Vasodilatatoren einnehmen.„Es liegt auch in der Verantwortung derjenigen, die LHD behandeln, pulmonale Vasodilatatoren nicht routinemäßig zu verschreiben, so verlockend das auch sein mag, da wir Anzeichen für Schäden durch diese Medikamente bei Patienten mit PH gesehen haben“, sagt er.Ein umfassender Bericht einer Arbeitsgruppe des 7. Weltsymposiums über pulmonale Hypertonie, bei dem Maron als Vorsitzender und Erstautor fungierte, identifizierte eine grundlegende Einschränkung der aktuellen Ansätze zur PH-LHD: die Tendenz, ein „Einheitsklassifizierungssystem“ auf sehr unterschiedliche Herzpathologien anzuwenden.Diese grobe Kategorisierung erfasst nicht die einzigartige Pathophysiologie, Prävalenzmuster, Prognose und das Ansprechen auf die Behandlung, die spezifische LHD-Subtypen kennzeichnen. Maron et al. argumentieren, dass die Aufschlüsselung von PH-LHD in distinkte klinische Entitäten einen entscheidenden Schritt hin zu personalisierter Medizin und verbesserten Patientenergebnissen darstellt.Die therapeutischen Implikationen der Disaggregation reichen über einfache Aspekte hinaus und umfassen grundlegende Unterschiede in den Behandlungsansätzen und den zu erwartenden Ergebnissen.Für eine definitive PH-Diagnose ist eine Rechtsherzkatheteruntersuchung erforderlich, die laut Ilonze in etwa einem Viertel der Fälle ausgelassen wird. Paradoxerweise wird PH-LHD gelegentlich überdiagnostiziert, da bei manchen Patienten die Diagnose PH-LHD allein auf echokardiografischen Schätzungen basiert, ohne dass eine adäquate hämodynamische Bestätigung erfolgt.Die zunehmende Bedeutung von SGLT-2-Inhibitoren verdeutlicht, wie hilfreich eine Disaggregation sein könnte. Obwohl diese Medikamente bei Herzinsuffizienzpatienten durch ihre Wirkung auf das Plasmavolumen und Entzündungsprozesse vielversprechend sind, bleibt ihre Rolle bei Herzklappenerkrankungen oder spezifischen Kardiomyopathien unklar und erfordert möglicherweise subtypspezifische Untersuchungen.Zu viele Patienten, zu wenige Spezialisten„Die schiere Anzahl der PH-LHD-Fälle ist überwältigend“, sagt Ilonze. „Die PH-Spezialisten können derzeit nicht alle Patienten behandeln, die unsere Hilfe benötigen. Daher ist es umso wichtiger, dass Kardiologen ein hohes Maß an Aufmerksamkeit für PH entwickeln und besser für deren Behandlung gerüstet sind.“Er empfiehlt ein Modell der gemeinsamen Betreuung. „Wenn ein Patient an PH-LHD leidet und keine spezifische Lungentherapie benötigt, kann eine gemeinsame Betreuung erfolgen, bei der der Patient vom örtlichen Kardiologen betreut wird, aber vielleicht jährliche Kontrolluntersuchungen bei einem Spezialisten wahrnimmt.“Herzinsuffizienzspezialisten sind für diese Bemühungen von entscheidender Bedeutung, nicht zuletzt, weil viele Patienten mit pulmonaler Hypertonie und Linksherzinsuffizienz (PH-LHD) an einer langjährigen Kardiomyopathie leiden. Sie spielen auch eine wesentliche Rolle bei der Beurteilung der Eignung für eine Herztransplantation, der Optimierung der leitliniengerechten medikamentösen Therapie und der Erkennung von Fällen, in denen der pulmonale Druck das Eingriffs- und Operationsrisiko signifikant erhöht. Patienten mit einer schweren Kardiomyopathie und einer schweren pulmonalen Hypertonie (PH) kommen aufgrund der hohen perioperativen Mortalität nicht mehr für eine Herztransplantation infrage.
Der gut belegte Zusammenhang zwischen COVID-19 und Hyperkoagulabilität sowie Lungenembolie hat Bedenken hinsichtlich eines möglichen Anstiegs der CTEPH-Inzidenz geweckt.<sup> 7,8</sup> Das Virus löst einen prothrombotischen Zustand aus, der über die akute Erkrankung hinaus anhalten kann; dokumentierte Fälle von Lungenembolie traten Wochen bis Monate nach der Erstinfektion auf.Obwohl die endgültigen epidemiologischen Daten zur Inzidenz von CTEPH nach COVID-19 noch ausstehen, ist der zeitliche Zusammenhang besorgniserregend. Da sich CTEPH typischerweise sechs Monate bis zwei Jahre nach einem initialen embolischen Ereignis entwickelt, stehen wir möglicherweise erst am Anfang der vollen kardiovaskulären Folgen der Höhepunkte der Pandemie.„Wir wissen es noch nicht genau, aber wir werden dies möglicherweise häufiger beobachten, wenn die Menschen einige Jahre länger von COVID-19 verschont bleiben. Wir gehen aber auch davon aus, dass CTEPH generell unterschätzt wird und dass es viele Menschen gibt, die möglicherweise an einer schlechten Atemwegsgesundheit leiden, dieses Narbengewebe haben und es einfach nicht wissen“, sagt Oliveros.Das Besondere an der chronisch-thromboembolischen pulmonalen Hypertonie (CTEPH) unter den PH-Subtypen ist die Möglichkeit der chirurgischen Heilung durch pulmonale Thromboendarterektomie (PTE). Bei diesem komplexen Eingriff, der in spezialisierten Zentren durchgeführt wird, werden organisierte Blutgerinnsel unter tiefer Hypothermie und Kreislaufstillstand entfernt.Die jüngsten Ergebnisse der PTE sind vielversprechend: Eine Metaanalyse aus dem Jahr 2025 berichtet von einer Mortalität von 8,4 % über alle Zentren hinweg, die in spezialisierten Zentren jedoch auf 1–2 % sinkt.⁶ UC San Diego Health, wo das Verfahren entwickelt wurde, hat bereits über 5.000 PTEs mit hervorragenden Ergebnissen durchgeführt.„Eine meiner Sorgen ist, dass einige Zentren, die nicht ausreichend auf die Behandlung dieser Erkrankung vorbereitet sind, versuchen könnten, das Blutgerinnsel zu entfernen, weil sie es für ein akutes, frisches Gerinnsel halten, es aber nicht vollständig entfernen. Sie entfernen möglicherweise nur einen Teil davon und verschließen die Wunde wieder“, bemerkt Oliveros. „Daher besteht bei dieser speziellen Patientengruppe Bedarf an multidisziplinär ausgebildeten Teams, bestehend aus einem Spezialisten für pulmonale Hypertonie, einem Radiologen, einem Chirurgen und einem interventionellen Kardiologen, die alle in der Diagnose und Behandlung dieser spezifischen Erkrankung geschult sind.“Für Patienten, die für eine PTE nicht geeignet sind, stellen die Ballon-Pulmonalangioplastie und die gezielte medikamentöse Therapie mit Riociguat sinnvolle Alternativen dar.Die COVID-19-Pandemie unterstreicht die Bedeutung der Aufklärung über CTEPH. Die Herausforderung besteht in der Erkennung: Viele COVID-bedingte embolische Ereignisse verlaufen möglicherweise subklinisch, und der schleichende Beginn von CTEPH-Symptomen – fortschreitende Atemnot, Müdigkeit und Belastungsintoleranz – kann leicht fälschlicherweise als „Long COVID“ oder Dekonditionierung interpretiert werden.„Manche Menschen haben ausgedehnte chronische Blutgerinnsel in der Lunge, ohne es zu wissen, sind aber nicht in der Lage, einen Raum zu durchqueren. Wir führen eine PTE durch und entfernen das gesamte Gerinnsel, woraufhin sich ihr Zustand dramatisch verbessert“, sagt Oliveros.„Da die Menschen immer älter werden, steigt auch die Wahrscheinlichkeit für Erkrankungen des linken Herzens, sei es durch Kardiomyopathie, Herzinsuffizienz oder Herzklappenerkrankungen, was wiederum das Risiko für pulmonale Hypertonie erhöht“, sagt Ilonze, Erstautor eines kürzlich in JACC: Heart Failure veröffentlichten Übersichtsartikels zum Thema pulmonale Hypertonie bei Linksherzerkrankungen.⁵Er vermutet, dass Ärzte nicht ausreichend darüber nachdenken, ob ihre Patienten mit Linksherzpathologie möglicherweise auch an pulmonaler Hypertonie (PH) leiden. Symptome der PH in Verbindung mit einer Linksherzerkrankung (Atemnot, Müdigkeit, Belastungsintoleranz) werden oft ausschließlich der zugrunde liegenden Linksherzerkrankung zugeschrieben, wobei Ärzte annehmen, dass sich verschlimmernde Symptome eher auf das Fortschreiten der Herzinsuffizienz als auf die Entwicklung einer begleitenden PH in Verbindung mit einer Linksherzerkrankung zurückzuführen sind.„Ein neues Medikament gegen PAH zu haben, ist aufregend, aber es verschärft das Problem wahrscheinlich noch etwas. Es besteht immer die Tendenz, nach der Krankheit zu suchen, für die es ein vielversprechendes neues Medikament gibt, und nicht nach der Krankheit, für die man dem Patienten nichts Besonderes anbieten kann.“Ein weiteres mögliches Hindernis für eine korrekte Diagnose ist der therapeutische Nihilismus: Da pulmonale Vasodilatatoren bei PH-LHD kontraindiziert sind und das neu zugelassene und äußerst wirksame Sotatercept nur für PAH zugelassen ist, könnten einige Kliniker der Ansicht sein, dass der therapeutische Nutzen einer spezifischen Diagnose begrenzt ist, was zu einer weniger aggressiven Diagnostik führt.Wenn bei Patienten bereits eine Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF), Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) oder eine Herzklappenerkrankung diagnostiziert wurde, besteht weniger Anreiz für eine invasive hämodynamische Untersuchung. „Die Denkweise ändert sich: ‚Wir wissen ja bereits, dass sie eine Herzerkrankung haben‘, anstatt die pulmonale Gefäßkomponente zu untersuchen“, erklärt Ilonze. Dies führt dazu, dass die pulmonale Hypertonie in Kombination mit einer Linksherzerkrankung (LHD) unerkannt und unbehandelt bleibt. „Es ist wichtig zu erkennen, dass die Entwicklung einer pulmonalen Hypertonie zusätzlich zur LHD die Prognose der bereits bestehenden LHD verschlechtert“, fügt er hinzu.Maron weist darauf hin, dass es zwei Patientengruppen gibt, bei denen eine „strenge Vorgehensweise bei pulmonaler Hypertonie“ besonders wichtig ist. Die erste Gruppe umfasst Patienten mit Mitralklappenerkrankung, bei denen das Vorliegen einer pulmonalen Hypertonie aufgrund der Erkrankung eine Indikation für eine Operation darstellen kann, und die zweite Gruppe betrifft Patienten, die Vasodilatatoren einnehmen.„Es liegt auch in der Verantwortung derjenigen, die LHD behandeln, pulmonale Vasodilatatoren nicht routinemäßig zu verschreiben, so verlockend das auch sein mag, da wir Anzeichen für Schäden durch diese Medikamente bei Patienten mit PH gesehen haben“, sagt er.Ein umfassender Bericht einer Arbeitsgruppe des 7. Weltsymposiums über pulmonale Hypertonie, bei dem Maron als Vorsitzender und Erstautor fungierte, identifizierte eine grundlegende Einschränkung der aktuellen Ansätze zur PH-LHD: die Tendenz, ein „Einheitsklassifizierungssystem“ auf sehr unterschiedliche Herzpathologien anzuwenden.Diese grobe Kategorisierung erfasst nicht die einzigartige Pathophysiologie, Prävalenzmuster, Prognose und das Ansprechen auf die Behandlung, die spezifische LHD-Subtypen kennzeichnen. Maron et al. argumentieren, dass die Aufschlüsselung von PH-LHD in distinkte klinische Entitäten einen entscheidenden Schritt hin zu personalisierter Medizin und verbesserten Patientenergebnissen darstellt.Die therapeutischen Implikationen der Disaggregation reichen über einfache Aspekte hinaus und umfassen grundlegende Unterschiede in den Behandlungsansätzen und den zu erwartenden Ergebnissen.Für eine definitive PH-Diagnose ist eine Rechtsherzkatheteruntersuchung erforderlich, die laut Ilonze in etwa einem Viertel der Fälle ausgelassen wird. Paradoxerweise wird PH-LHD gelegentlich überdiagnostiziert, da bei manchen Patienten die Diagnose PH-LHD allein auf echokardiografischen Schätzungen basiert, ohne dass eine adäquate hämodynamische Bestätigung erfolgt.Die zunehmende Bedeutung von SGLT-2-Inhibitoren verdeutlicht, wie hilfreich eine Disaggregation sein könnte. Obwohl diese Medikamente bei Herzinsuffizienzpatienten durch ihre Wirkung auf das Plasmavolumen und Entzündungsprozesse vielversprechend sind, bleibt ihre Rolle bei Herzklappenerkrankungen oder spezifischen Kardiomyopathien unklar und erfordert möglicherweise subtypspezifische Untersuchungen.Zu viele Patienten, zu wenige Spezialisten„Die schiere Anzahl der PH-LHD-Fälle ist überwältigend“, sagt Ilonze. „Die PH-Spezialisten können derzeit nicht alle Patienten behandeln, die unsere Hilfe benötigen. Daher ist es umso wichtiger, dass Kardiologen ein hohes Maß an Aufmerksamkeit für PH entwickeln und besser für deren Behandlung gerüstet sind.“Er empfiehlt ein Modell der gemeinsamen Betreuung. „Wenn ein Patient an PH-LHD leidet und keine spezifische Lungentherapie benötigt, kann eine gemeinsame Betreuung erfolgen, bei der der Patient vom örtlichen Kardiologen betreut wird, aber vielleicht jährliche Kontrolluntersuchungen bei einem Spezialisten wahrnimmt.“Herzinsuffizienzspezialisten sind für diese Bemühungen von entscheidender Bedeutung, nicht zuletzt, weil viele Patienten mit pulmonaler Hypertonie und Linksherzinsuffizienz (PH-LHD) an einer langjährigen Kardiomyopathie leiden. Sie spielen auch eine wesentliche Rolle bei der Beurteilung der Eignung für eine Herztransplantation, der Optimierung der leitliniengerechten medikamentösen Therapie und der Erkennung von Fällen, in denen der pulmonale Druck das Eingriffs- und Operationsrisiko signifikant erhöht. Patienten mit einer schweren Kardiomyopathie und einer schweren pulmonalen Hypertonie (PH) kommen aufgrund der hohen perioperativen Mortalität nicht mehr für eine Herztransplantation infrage.
 PH-LHD ist in jeder kardiologischen Praxis weit verbreitet, ob erkannt oder nicht, so Ilonze. Jeder Kardiologe sollte die Fähigkeit zur Erkennung entwickeln, einen klaren Überweisungsweg für komplexe Fälle haben und verstehen, dass erhöhter pulmonaler Druck ein höheres Risikoprofil darstellt, das die klinische Entscheidungsfindung im gesamten Spektrum der kardiovaskulären Versorgung beeinflussen sollte.Der Weg nach vornSotatercept verändert die Situation nicht nur bei PAH, sondern auch im gesamten Fachgebiet. Die Erkrankung wird weiterhin unterdiagnostiziert, doch die Einführung dieser bahnbrechenden Therapie stellt höhere Anforderungen an Kliniker, Wissenslücken in der Beurteilung des rechten Herzens zu schließen, sich mit der PH-Klassifikation und Hämodynamik vertraut zu machen und klarere Überweisungswege zu PH-Spezialisten zu schaffen.Die Früherkennung vor dem Auftreten irreversibler Veränderungen, die Ausweitung der Anwendung krankheitsmodifizierender Therapien im gesamten Spektrum der pulmonalen Hypertonie und die fortgesetzte Forschung an Ansätzen, die auf die grundlegende Pathophysiologie und nicht nur auf die Symptome abzielen, stellen die Zukunft der Behandlung pulmonaler Hypertonie dar.Dieser Artikel wurde von Debra L Beck, MSc , verfasst .Referenzen
PH-LHD ist in jeder kardiologischen Praxis weit verbreitet, ob erkannt oder nicht, so Ilonze. Jeder Kardiologe sollte die Fähigkeit zur Erkennung entwickeln, einen klaren Überweisungsweg für komplexe Fälle haben und verstehen, dass erhöhter pulmonaler Druck ein höheres Risikoprofil darstellt, das die klinische Entscheidungsfindung im gesamten Spektrum der kardiovaskulären Versorgung beeinflussen sollte.Der Weg nach vornSotatercept verändert die Situation nicht nur bei PAH, sondern auch im gesamten Fachgebiet. Die Erkrankung wird weiterhin unterdiagnostiziert, doch die Einführung dieser bahnbrechenden Therapie stellt höhere Anforderungen an Kliniker, Wissenslücken in der Beurteilung des rechten Herzens zu schließen, sich mit der PH-Klassifikation und Hämodynamik vertraut zu machen und klarere Überweisungswege zu PH-Spezialisten zu schaffen.Die Früherkennung vor dem Auftreten irreversibler Veränderungen, die Ausweitung der Anwendung krankheitsmodifizierender Therapien im gesamten Spektrum der pulmonalen Hypertonie und die fortgesetzte Forschung an Ansätzen, die auf die grundlegende Pathophysiologie und nicht nur auf die Symptome abzielen, stellen die Zukunft der Behandlung pulmonaler Hypertonie dar.Dieser Artikel wurde von Debra L Beck, MSc , verfasst .Referenzen
[list=decimal]
[*]Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase-3-Studie mit Sotatercept zur Behandlung der pulmonalen arteriellen Hypertonie. New Engl J Med 2023;388:1478-90.
[*]McLaughlin VV, Hoeper MM, Badesch DB, et al., für die HYPERION-Studienautoren. Sotatercept zur Behandlung der pulmonalen arteriellen Hypertonie im ersten Jahr nach der Diagnose. NEJM 2025; Online veröffentlicht am 30. September: DOI: 10.1056/NEJMoa2508170.
[*]Humbert M, McLaughlin VV, Badesch DB, et al. Sotatercept bei Patienten mit pulmonaler arterieller Hypertonie und hohem Sterberisiko. N Engl J Med 2025;392:1987-2000.
[*]Maron BA. Sotatercept und die klinische Transformation der pulmonalen arteriellen Hypertonie. N Engl J Med 2025;392:2059-61.
[*]Ilonze OJ, Ebong IA, Guglin M, et al. Überlegungen zur Diagnose und Behandlung der mit einer Linksherzerkrankung assoziierten pulmonalen Hypertonie. JACC: Heart Fail 2024;12:1328-42.
[*]Madani MM, Wiedenroth CB, Jenkins DP, Fadel E, de Perrot M. Pulmonale Thromboendarterektomie: die potenziell kurative Therapie der Wahl bei chronischer thromboembolischer pulmonaler Hypertonie. Ann Thorac Surg 2025;119:756-67.
[*]de Jong CMM, Visser C, Bemelmans RHH, et al. Chronische thromboembolische pulmonale Hypertonie und Auflösung des Thrombus nach COVID-19-assoziierter Lungenembolie. Eur Respir J 2023;61:2300171.
[*]Reddy SA, Newman J, Leavy OC, et al. CTEPH ist eine seltene Komplikation von COVID-19: Nationale Überwachungs- und Beobachtungskohortenstudien aus Großbritannien. Eur Respir J 2024;64:2301742.
[/list]
[list=decimal]
[*]Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase-3-Studie mit Sotatercept zur Behandlung der pulmonalen arteriellen Hypertonie. New Engl J Med 2023;388:1478-90.
[*]McLaughlin VV, Hoeper MM, Badesch DB, et al., für die HYPERION-Studienautoren. Sotatercept zur Behandlung der pulmonalen arteriellen Hypertonie im ersten Jahr nach der Diagnose. NEJM 2025; Online veröffentlicht am 30. September: DOI: 10.1056/NEJMoa2508170.
[*]Humbert M, McLaughlin VV, Badesch DB, et al. Sotatercept bei Patienten mit pulmonaler arterieller Hypertonie und hohem Sterberisiko. N Engl J Med 2025;392:1987-2000.
[*]Maron BA. Sotatercept und die klinische Transformation der pulmonalen arteriellen Hypertonie. N Engl J Med 2025;392:2059-61.
[*]Ilonze OJ, Ebong IA, Guglin M, et al. Überlegungen zur Diagnose und Behandlung der mit einer Linksherzerkrankung assoziierten pulmonalen Hypertonie. JACC: Heart Fail 2024;12:1328-42.
[*]Madani MM, Wiedenroth CB, Jenkins DP, Fadel E, de Perrot M. Pulmonale Thromboendarterektomie: die potenziell kurative Therapie der Wahl bei chronischer thromboembolischer pulmonaler Hypertonie. Ann Thorac Surg 2025;119:756-67.
[*]de Jong CMM, Visser C, Bemelmans RHH, et al. Chronische thromboembolische pulmonale Hypertonie und Auflösung des Thrombus nach COVID-19-assoziierter Lungenembolie. Eur Respir J 2023;61:2300171.
[*]Reddy SA, Newman J, Leavy OC, et al. CTEPH ist eine seltene Komplikation von COVID-19: Nationale Überwachungs- und Beobachtungskohortenstudien aus Großbritannien. Eur Respir J 2024;64:2301742.
[/list]
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.