
- Beiträge: 1757
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Nutzung von NLRX1: Ein neuer Ansatz zur Linderung von Entzündungen bei PH
Nutzung von NLRX1: Ein neuer Ansatz zur Linderung von Entzündungen bei PH
08 Nov 2025 16:19 #2432
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Nutzung von NLRX1: Ein neuer Ansatz zur Linderung von Entzündungen bei PH wurde erstellt von danny
respiratory-research.biomedcentral.com/a...1-025-03364-wNutzung von NLRX1: Ein neuer Ansatz zur Linderung von Entzündungen bei pulmonaler Hypertonie
") zu einer Hochregulation von Entzündungsmediatoren und fördert gleichzeitig oxidativen Stress sowie die Aktivierung des nukleären Faktors E2-verwandten Faktors 2 (Nrf2). Der Aktivator NX-13 von NLRX1 kann die Produktion von entzündungsfördernden Zytokinen in durch Hypoxie stimulierten BMDMs reduzieren und oxidativen Stress mindern.SchlussfolgerungenNLRX1 kann durch seine entzündungshemmenden und antioxidativen Eigenschaften hypoxiebedingte Lungenentzündungen unterdrücken und hypoxiebedingte pulmonale Hypertonie lindern. Daher könnte die gezielte Hochregulierung von NLRX1 eine neue Behandlungsstrategie für HPH darstellen.HintergrundDie hypoxische pulmonale Hypertonie (HPH) ist eine Erkrankung, die durch einen erhöhten pulmonalen Arteriendruck aufgrund einer längeren Exposition gegenüber einer hypoxischen Umgebung gekennzeichnet ist. Sie tritt häufig bei Personen auf, die in großen Höhen leben, und bei Patienten mit chronisch-obstruktiver Lungenerkrankung (COPD) [
1
]. Die pulmonale Gefäßremodellierung gilt allgemein als Hauptursache für den erhöhten pulmonalen Arteriendruck, während Hypoxie als zentraler Auslöser der HPH angesehen wird, da sie eine Entzündungsreaktion im Immunsystem stimuliert. Anhaltende Hypoxie induziert die Proliferation, Migration und phänotypische Veränderung von glatten Muskelzellen der Pulmonalarterie (PASMCs), was zu einer Verdickung der Gefäßwand und einer Verengung des Lumens führt [
2
]. Neuere Erkenntnisse deuten darauf hin, dass hypoxieinduzierte Entzündungen und metabolische Dysregulationen in Gefäßzellen zur Entwicklung der HPH beitragen. Perivaskuläre Entzündungen sind eng mit der Verdickung der Intima und Media der Gefäßwand verbunden [
3
]. Histopathologische Untersuchungen zeigen bei Patienten mit HPH entzündliche Infiltrate aus Makrophagen, Mastzellen und Neutrophilen, die pulmonale Gefäßläsionen umgeben [
4
]. Immunzellen interagieren nicht nur direkt mit pulmonalen Gefäßzellen, sondern setzen auch Entzündungsmediatoren frei, die die Proliferation von PASMCs fördern oder eine Endothelzellfunktionsstörung induzieren. Diese Prozesse treiben gemeinsam das Gefäßremodeling voran und führen zu einem erhöhten pulmonalen Arteriendruck.Die Familie der Nod-ähnlichen Rezeptoren (NLR) ist eine wichtige Gruppe von Mustererkennungsrezeptoren im menschlichen Körper, die primär die angeborene Immunantwort steuern und die innere Homöostase aufrechterhalten [
5
]. Unter diesen Rezeptoren nimmt NLRX1 eine herausragende Stellung ein. Er ist vorwiegend im Zytoplasma und in den Mitochondrien lokalisiert und spielt dort eine zentrale Rolle bei der Regulation von Immunität, Antioxidation und Apoptose [
6
]. Studien haben gezeigt, dass NLRX1 die Lipopolysaccharid-induzierte NF-κB-Aktivierung nachgeschaltet von Toll-like-Rezeptor 4 (TLR4) negativ reguliert, indem es in Fibroblasten oder Makrophagen mit dem TNF-Rezeptor-assoziierten Faktor 6 (TRAF6) interagiert und einen Komplex bildet [
7
]. In der T-Zell-Immunantwort moduliert NLRX1 die T-Zell-Aktivierung und -Differenzierung durch Beeinflussung der NF-κB-Signalübertragung [
8
,
9
]. NLRX1 besitzt bemerkenswerterweise eine Doppelfunktion in der Entzündungsreaktion und Immunregulation [
10
]. Es fördert Autophagie und Apoptose, um pathogene Mikroorganismen und Tumorzellen zu bekämpfen, und hemmt gleichzeitig Entzündungsprozesse, um Entzündungen zu lindern. Die zentrale Rolle von NLRX1 zeigt sich in der Pathogenese und Progression verschiedener Erkrankungen, darunter Virusinfektionen, Tumore und Autoimmunerkrankungen [
11
,
12
,
13
,
14
,
15
]. Obwohl die genaue Beteiligung von NLRX1 an der hypoxiebedingten pulmonalen Hypertonie (HPH) noch nicht vollständig erforscht ist, deuten bestehende Studien darauf hin, dass eine NLRX1-Supplementierung die Lungenentzündung deutlich reduzieren kann, wobei eine inverse Korrelation mit der Expressionsstärke entzündungsfördernder Zytokine besteht [
16
,
17
]. Diese Beobachtungen legen eine mögliche Rolle von NLRX1 bei der Linderung der hypoxiebedingten pulmonalen Hypertonie durch die Unterdrückung der Entzündungsreaktion im Lungengewebe nahe.Zusammenfassend lässt sich sagen, dass NLRX1 als Suppressor von NF-κB fungiert, einem zentralen Signalweg bei hypoxieinduzierter Entzündung und oxidativem Stress. Es wurde berichtet, dass NLRX1 die NF-κB-Signalübertragung in verschiedenen Zelltypen hemmt, was darauf hindeutet, dass seine entzündungshemmenden und antioxidativen Eigenschaften durch hypoxische Bedingungen beeinflusst werden könnten. Daher sind weitere Untersuchungen erforderlich, um den genauen Beitrag von NLRX1 im Kontext der Hypoxie-induzierten Hypoxie (HPH) aufzuklären.MethodenVersuchstiereWildtyp-Mäuse (WT) des Stammes C57BL/6 wurden vom Tierzentrum der Vierten Militärmedizinischen Universität (China) bezogen. NLRX1-Knockout-Mäuse (NLRX1 <sup>−/−</sup> ) des Stammes C57BL/6 wurden in Zusammenarbeit mit dem Institut für Mikrobiologie der Vierten Militärmedizinischen Universität generiert [
18
]. Beide Mäusestämme waren männlich und 6–8 Wochen alt. Die Protokolle dieser Studie wurden von der Ethikkommission für Tierversuche der Vierten Militärmedizinischen Universität genehmigt.Modellierung und Gruppierung von PH-MäusenWT- und NLRX1<sup> −/−</sup> -Mäuse wurden randomisiert in vier Gruppen eingeteilt: Kontrollgruppe, NLRX1<sup> −/− </sup>-Gruppe , Hypoxiegruppe und Hypoxie + NLRX1<sup> −/−</sup> -Gruppe (jeweils 20 Mäuse). Die Mäuse wurden für sechs Wochen in einer Unterdruckkammer (Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> , 10 % O <sub>2 </sub> ) oder unter Raumluftbedingungen (Kontrollgruppe, NLRX1 <sup>−/−</sup> , 21 % O <sub> 2</sub>) gehalten. Während der gesamten Studie hatten alle Mäuse sowohl unter Hypoxie- als auch unter Normoxiebedingungen uneingeschränkten Zugang zu Futter und Wasser.Hämodynamik und HypertrophiemessungDer systolische Druck im rechten Ventrikel (RVSP) von Mäusen wurde nach Pentobarbital-Natrium-Narkose gemessen. Die Rippen wurden chirurgisch freigelegt, und der Druckwandler wurde nach Anschluss an die intravenöse Nadel in den rechten Ventrikel kanüliert. Der Druck im rechten Ventrikel wurde mindestens 30 Sekunden lang kontinuierlich gemessen [
19
]. Alle Daten wurden mit dem Datenerfassungssystem Powerlab (AD Instruments) aufgezeichnet und analysiert. Im Anschluss an die Datenerfassung wurden die Mäuse durch sofortiges Ausbluten euthanasiert, und Lungen- und Herzgewebe wurden entnommen. Der rechte Ventrikel (RV) wurde vom linken Ventrikel (LV) einschließlich der Septumwand (S) getrennt, und beide wurden gewogen, um das Verhältnis des Gewichts der freien Wand des rechten Ventrikels zum Gewicht des linken Ventrikels plus Septum (RV/LV + S) zu berechnen [
20
].Histopathologische Untersuchung des rechten Ventrikels und der LungeDie aus den Mäusen entnommenen Lungen- und Herzgewebe wurden in Paraformaldehyd eingelegt und 24 Stunden lang fixiert. Anschließend wurden diese Gewebe in Wachsblöcke eingebettet, aus denen 5 μm dicke Schnitte angefertigt wurden. Die Gewebeschnitte wurden nach Hämatoxylin-Eosin-Färbung mikroskopisch untersucht. Die Gefäßwanddicke (WT), der äußere Gefäßdurchmesser (ED), die Gefäßwandfläche (WA) und die gesamte Gefäßfläche (TA) wurden gemessen, um WT% (WT/ED) und WA% (WA/TA) zu berechnen [21
]
. Die Dicke des rechten Ventrikels wurde mit einem Olympus-System gemessen.ImmunfluoreszenzDie Lungenschnitte wurden zunächst dreimal mit PBS gespült und anschließend durch Einlegen in Triton X-100 permeabilisiert. Nach Blockierung mit einer Immunfluoreszenz-Blockierungslösung (P0102, Beyotime) wurden die Schnitte über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-CD4 (ab133616, 1:500, Abcam), anti-F4/80 (29414-1-AP, 1:200, Proteintech), anti-α-SMA (A17910, 1:200, Abclonal) und anti-Nrf2 (A11159, 1:100, Abclonal). Anschließend erfolgte eine einstündige Inkubation mit sekundären Fluoreszenz-Antikörpern (ab150077, 1:500, ab150115, 1:200, Abcam) und eine 30-minütige Inkubation mit DAPI (C1005, Beyotime). Die Fluoreszenz wurde mittels Fluoreszenzmikroskopie abgebildet und die Daten mit Caseviewer verarbeitet.Isolierung und Kultivierung von aus dem Knochenmark stammenden Makrophagen (BMDMs)Für die Isolierung von Knochenmarkzellen wurden WT- und NLRX1 −/−- Mäuse im Alter von 4 bis 6 Wochen ausgewählt [
22
]. Nach der Erythrozytenlyse wurden die extrahierten Knochenmarkzellen in 2 ml PBS resuspendiert. Anschließend wurden die Zellen in RPMI 1640 mit 10 % FBS (10099-141, Gibco) und einer Konzentration von 10 ng/ml m-CSF (HZ-1192, Proteintech) überführt. Nach 24-stündiger Inkubation wurde die Suspension in 6-Well-Platten pipettiert. Die Proliferation und Differenzierung der BMDMs erstreckte sich über eine Woche, sodass das Medium an den Tagen 2, 4 und 6 erneuert werden musste.Zellbehandlung und Gruppierung von BMDMsWT- und NLRX1<sup> −/−</sup> -BMDMs wurden randomisiert in fünf Gruppen eingeteilt: Kontrolle, NLRX1 <sup>−/−</sup> , Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> und Hypoxie + NX-13. NX-13 (HY-141521, MedChemExpress) wurde in DMSO gelöst und auf eine Endkonzentration von 0,5 µM eingestellt [
8
,
23
]. Die Hypoxie-Gruppen wurden 24–48 h lang hypoxisch in einem Inkubator mit 5 % O <sub>2</sub> , die Normoxie-Gruppen in einem Inkubator mit 21 % O <sub>2 </sub> kultiviert . Anschließend wurden Zellproben für weitere Experimente entnommen.RNA-Extraktion und -NachweisRNA wurde aus Lungengewebe und BMDMs mit dem RNA-Extraktionskit (R0026, Beyotime) isoliert. Die RNA-Konzentration wurde gemessen und verdünnt. Anschließend erfolgte die cDNA-Synthese mit dem SweScript RT cDNA-Synthesekit (G3333, Servicebio). Das TB Green PCR-Kit (G3320, Servicebio) wurde den Proben zugegeben. Die quantitative Echtzeit-PCR (qRT-PCR) wurde auf einem PCR-Gerät von Biosystems durchgeführt. Die Genexpression wurde im Vergleich zu den Kontrollen mit der 2<sup>-ΔΔCt</sup>-Formel analysiert. Die Expression wurde auf die mittlere Expression aller Kontrollprobanden normiert.Folgende Primer wurden verwendet: β-Actin, 5'-CACGATGGAGGGGCCGGACTCATC-3' (vorwärts) und 5' -TAAAGACCTCTATGCCAACACAGT-3' (rückwärts); NLRX1, 5'-CAGATTGGTAACAAAGGAGCCA-3' (vorwärts) und 5'-CGTTCGGTTTATCTTCAGAGCA-3' (rückwärts).Western BlotGesamtproteine wurden aus Lungengewebe und BMDMs isoliert und die Konzentration mittels BCA-Assay-Kit (P0012, Beyotime) quantifiziert. Proben mit 50 µg Protein wurden auf ein 10%iges SDS-PAGE-Gel aufgetragen und einer einstündigen Gelelektrophorese unterzogen. Anschließend wurden die Proteine auf PVDF-Membranen transferiert und mit Western-Blot-Blockierungslösung (P30500, NCM Biotech) behandelt. Die Membranen wurden über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-NLRX1 (17215-1-AP, 1:1000, Proteintech), anti-IL-1β (A16288, 1:1000, Abclonal), anti-IL-18 (A1115, 1:1000, Abclonal), anti-pNF-κB p65 (82335-1-RR, 1:1000, Proteintech), anti-NF-κB p65 (10745-1-AP, 1:1000, Proteintech), anti-Nrf2 (A0674, 1:1000, Abclonal), anti-Keap1 (10503-2-AP, 1:1000, Proteintech) und anti-β-Actin (ab8227, 1:5000, Abcam). Inkubation mit dem Sekundärantikörper (RGAR001, 1:5000, Proteintech) für 1 h. Der Nachweis erfolgte mittels Chemilumineszenzsystem (CLINX ChemiScope).ZytokinmessungNach Aspiration und Zentrifugation des Überstands aus der BMDM-Kultur wurde die Konzentration von IL-1β und IL-6 mittels ELISA-Kits (R&D) bestimmt. Die Gebrauchsanweisung wurde vom Hersteller bereitgestellt.Messung der Superoxiddismutase (SOD)-AktivitätDie Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die Gesamt-SOD-Aktivität im Überstand mithilfe des Gesamt-SOD-Assay-Kits (S0101S, Beyotime) gemäß den Herstellerangaben bestimmt.Messung der gesamten antioxidativen Kapazität (TAC)Die Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die antioxidative Gesamtkapazität (TAC) im Überstand mithilfe des Testkits zur Bestimmung der antioxidativen Gesamtkapazität (S0121, Beyotime) gemäß den Herstellerangaben bestimmt.Statistische AnalysenDie Ergebnisse wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt und mit GraphPad Prism 9.0 analysiert. Signifikante Unterschiede zwischen den beiden Gruppen wurden mittels t-Test nach Student ermittelt. Unterschiede zwischen mehreren Gruppen wurden durch eine einfaktorielle Varianzanalyse (ANOVA) mit gegebenenfalls anschließendem Bonferroni-Post-hoc-Test untersucht. Ein p-Wert < 0,05 wurde als statistisch signifikant angesehen.ErgebnisseHypoxie führte zu einer verminderten Expression von NLRX1 im Lungengewebe von Mäusen.NLRX1 ist für seine negative Regulation von Entzündungen und Oxidation bekannt. Der Einfluss von Hypoxie auf die NLRX1-Funktion wurde jedoch bisher nicht untersucht. Daher analysierten wir die NLRX1-Expression im Lungengewebe von Mäusen mit HPH. Unsere Ergebnisse (Abb.
1
A und zeigen eine Reduktion sowohl der mRNA- als auch der Proteinspiegel von NLRX1 im Lungengewebe von HPH-Mäusen im Vergleich zur Kontrollgruppe.Abb. 1
zu einer Hochregulation von Entzündungsmediatoren und fördert gleichzeitig oxidativen Stress sowie die Aktivierung des nukleären Faktors E2-verwandten Faktors 2 (Nrf2). Der Aktivator NX-13 von NLRX1 kann die Produktion von entzündungsfördernden Zytokinen in durch Hypoxie stimulierten BMDMs reduzieren und oxidativen Stress mindern.SchlussfolgerungenNLRX1 kann durch seine entzündungshemmenden und antioxidativen Eigenschaften hypoxiebedingte Lungenentzündungen unterdrücken und hypoxiebedingte pulmonale Hypertonie lindern. Daher könnte die gezielte Hochregulierung von NLRX1 eine neue Behandlungsstrategie für HPH darstellen.HintergrundDie hypoxische pulmonale Hypertonie (HPH) ist eine Erkrankung, die durch einen erhöhten pulmonalen Arteriendruck aufgrund einer längeren Exposition gegenüber einer hypoxischen Umgebung gekennzeichnet ist. Sie tritt häufig bei Personen auf, die in großen Höhen leben, und bei Patienten mit chronisch-obstruktiver Lungenerkrankung (COPD) [
1
]. Die pulmonale Gefäßremodellierung gilt allgemein als Hauptursache für den erhöhten pulmonalen Arteriendruck, während Hypoxie als zentraler Auslöser der HPH angesehen wird, da sie eine Entzündungsreaktion im Immunsystem stimuliert. Anhaltende Hypoxie induziert die Proliferation, Migration und phänotypische Veränderung von glatten Muskelzellen der Pulmonalarterie (PASMCs), was zu einer Verdickung der Gefäßwand und einer Verengung des Lumens führt [
2
]. Neuere Erkenntnisse deuten darauf hin, dass hypoxieinduzierte Entzündungen und metabolische Dysregulationen in Gefäßzellen zur Entwicklung der HPH beitragen. Perivaskuläre Entzündungen sind eng mit der Verdickung der Intima und Media der Gefäßwand verbunden [
3
]. Histopathologische Untersuchungen zeigen bei Patienten mit HPH entzündliche Infiltrate aus Makrophagen, Mastzellen und Neutrophilen, die pulmonale Gefäßläsionen umgeben [
4
]. Immunzellen interagieren nicht nur direkt mit pulmonalen Gefäßzellen, sondern setzen auch Entzündungsmediatoren frei, die die Proliferation von PASMCs fördern oder eine Endothelzellfunktionsstörung induzieren. Diese Prozesse treiben gemeinsam das Gefäßremodeling voran und führen zu einem erhöhten pulmonalen Arteriendruck.Die Familie der Nod-ähnlichen Rezeptoren (NLR) ist eine wichtige Gruppe von Mustererkennungsrezeptoren im menschlichen Körper, die primär die angeborene Immunantwort steuern und die innere Homöostase aufrechterhalten [
5
]. Unter diesen Rezeptoren nimmt NLRX1 eine herausragende Stellung ein. Er ist vorwiegend im Zytoplasma und in den Mitochondrien lokalisiert und spielt dort eine zentrale Rolle bei der Regulation von Immunität, Antioxidation und Apoptose [
6
]. Studien haben gezeigt, dass NLRX1 die Lipopolysaccharid-induzierte NF-κB-Aktivierung nachgeschaltet von Toll-like-Rezeptor 4 (TLR4) negativ reguliert, indem es in Fibroblasten oder Makrophagen mit dem TNF-Rezeptor-assoziierten Faktor 6 (TRAF6) interagiert und einen Komplex bildet [
7
]. In der T-Zell-Immunantwort moduliert NLRX1 die T-Zell-Aktivierung und -Differenzierung durch Beeinflussung der NF-κB-Signalübertragung [
8
,
9
]. NLRX1 besitzt bemerkenswerterweise eine Doppelfunktion in der Entzündungsreaktion und Immunregulation [
10
]. Es fördert Autophagie und Apoptose, um pathogene Mikroorganismen und Tumorzellen zu bekämpfen, und hemmt gleichzeitig Entzündungsprozesse, um Entzündungen zu lindern. Die zentrale Rolle von NLRX1 zeigt sich in der Pathogenese und Progression verschiedener Erkrankungen, darunter Virusinfektionen, Tumore und Autoimmunerkrankungen [
11
,
12
,
13
,
14
,
15
]. Obwohl die genaue Beteiligung von NLRX1 an der hypoxiebedingten pulmonalen Hypertonie (HPH) noch nicht vollständig erforscht ist, deuten bestehende Studien darauf hin, dass eine NLRX1-Supplementierung die Lungenentzündung deutlich reduzieren kann, wobei eine inverse Korrelation mit der Expressionsstärke entzündungsfördernder Zytokine besteht [
16
,
17
]. Diese Beobachtungen legen eine mögliche Rolle von NLRX1 bei der Linderung der hypoxiebedingten pulmonalen Hypertonie durch die Unterdrückung der Entzündungsreaktion im Lungengewebe nahe.Zusammenfassend lässt sich sagen, dass NLRX1 als Suppressor von NF-κB fungiert, einem zentralen Signalweg bei hypoxieinduzierter Entzündung und oxidativem Stress. Es wurde berichtet, dass NLRX1 die NF-κB-Signalübertragung in verschiedenen Zelltypen hemmt, was darauf hindeutet, dass seine entzündungshemmenden und antioxidativen Eigenschaften durch hypoxische Bedingungen beeinflusst werden könnten. Daher sind weitere Untersuchungen erforderlich, um den genauen Beitrag von NLRX1 im Kontext der Hypoxie-induzierten Hypoxie (HPH) aufzuklären.MethodenVersuchstiereWildtyp-Mäuse (WT) des Stammes C57BL/6 wurden vom Tierzentrum der Vierten Militärmedizinischen Universität (China) bezogen. NLRX1-Knockout-Mäuse (NLRX1 <sup>−/−</sup> ) des Stammes C57BL/6 wurden in Zusammenarbeit mit dem Institut für Mikrobiologie der Vierten Militärmedizinischen Universität generiert [
18
]. Beide Mäusestämme waren männlich und 6–8 Wochen alt. Die Protokolle dieser Studie wurden von der Ethikkommission für Tierversuche der Vierten Militärmedizinischen Universität genehmigt.Modellierung und Gruppierung von PH-MäusenWT- und NLRX1<sup> −/−</sup> -Mäuse wurden randomisiert in vier Gruppen eingeteilt: Kontrollgruppe, NLRX1<sup> −/− </sup>-Gruppe , Hypoxiegruppe und Hypoxie + NLRX1<sup> −/−</sup> -Gruppe (jeweils 20 Mäuse). Die Mäuse wurden für sechs Wochen in einer Unterdruckkammer (Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> , 10 % O <sub>2 </sub> ) oder unter Raumluftbedingungen (Kontrollgruppe, NLRX1 <sup>−/−</sup> , 21 % O <sub> 2</sub>) gehalten. Während der gesamten Studie hatten alle Mäuse sowohl unter Hypoxie- als auch unter Normoxiebedingungen uneingeschränkten Zugang zu Futter und Wasser.Hämodynamik und HypertrophiemessungDer systolische Druck im rechten Ventrikel (RVSP) von Mäusen wurde nach Pentobarbital-Natrium-Narkose gemessen. Die Rippen wurden chirurgisch freigelegt, und der Druckwandler wurde nach Anschluss an die intravenöse Nadel in den rechten Ventrikel kanüliert. Der Druck im rechten Ventrikel wurde mindestens 30 Sekunden lang kontinuierlich gemessen [
19
]. Alle Daten wurden mit dem Datenerfassungssystem Powerlab (AD Instruments) aufgezeichnet und analysiert. Im Anschluss an die Datenerfassung wurden die Mäuse durch sofortiges Ausbluten euthanasiert, und Lungen- und Herzgewebe wurden entnommen. Der rechte Ventrikel (RV) wurde vom linken Ventrikel (LV) einschließlich der Septumwand (S) getrennt, und beide wurden gewogen, um das Verhältnis des Gewichts der freien Wand des rechten Ventrikels zum Gewicht des linken Ventrikels plus Septum (RV/LV + S) zu berechnen [
20
].Histopathologische Untersuchung des rechten Ventrikels und der LungeDie aus den Mäusen entnommenen Lungen- und Herzgewebe wurden in Paraformaldehyd eingelegt und 24 Stunden lang fixiert. Anschließend wurden diese Gewebe in Wachsblöcke eingebettet, aus denen 5 μm dicke Schnitte angefertigt wurden. Die Gewebeschnitte wurden nach Hämatoxylin-Eosin-Färbung mikroskopisch untersucht. Die Gefäßwanddicke (WT), der äußere Gefäßdurchmesser (ED), die Gefäßwandfläche (WA) und die gesamte Gefäßfläche (TA) wurden gemessen, um WT% (WT/ED) und WA% (WA/TA) zu berechnen [21
]
. Die Dicke des rechten Ventrikels wurde mit einem Olympus-System gemessen.ImmunfluoreszenzDie Lungenschnitte wurden zunächst dreimal mit PBS gespült und anschließend durch Einlegen in Triton X-100 permeabilisiert. Nach Blockierung mit einer Immunfluoreszenz-Blockierungslösung (P0102, Beyotime) wurden die Schnitte über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-CD4 (ab133616, 1:500, Abcam), anti-F4/80 (29414-1-AP, 1:200, Proteintech), anti-α-SMA (A17910, 1:200, Abclonal) und anti-Nrf2 (A11159, 1:100, Abclonal). Anschließend erfolgte eine einstündige Inkubation mit sekundären Fluoreszenz-Antikörpern (ab150077, 1:500, ab150115, 1:200, Abcam) und eine 30-minütige Inkubation mit DAPI (C1005, Beyotime). Die Fluoreszenz wurde mittels Fluoreszenzmikroskopie abgebildet und die Daten mit Caseviewer verarbeitet.Isolierung und Kultivierung von aus dem Knochenmark stammenden Makrophagen (BMDMs)Für die Isolierung von Knochenmarkzellen wurden WT- und NLRX1 −/−- Mäuse im Alter von 4 bis 6 Wochen ausgewählt [
22
]. Nach der Erythrozytenlyse wurden die extrahierten Knochenmarkzellen in 2 ml PBS resuspendiert. Anschließend wurden die Zellen in RPMI 1640 mit 10 % FBS (10099-141, Gibco) und einer Konzentration von 10 ng/ml m-CSF (HZ-1192, Proteintech) überführt. Nach 24-stündiger Inkubation wurde die Suspension in 6-Well-Platten pipettiert. Die Proliferation und Differenzierung der BMDMs erstreckte sich über eine Woche, sodass das Medium an den Tagen 2, 4 und 6 erneuert werden musste.Zellbehandlung und Gruppierung von BMDMsWT- und NLRX1<sup> −/−</sup> -BMDMs wurden randomisiert in fünf Gruppen eingeteilt: Kontrolle, NLRX1 <sup>−/−</sup> , Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> und Hypoxie + NX-13. NX-13 (HY-141521, MedChemExpress) wurde in DMSO gelöst und auf eine Endkonzentration von 0,5 µM eingestellt [
8
,
23
]. Die Hypoxie-Gruppen wurden 24–48 h lang hypoxisch in einem Inkubator mit 5 % O <sub>2</sub> , die Normoxie-Gruppen in einem Inkubator mit 21 % O <sub>2 </sub> kultiviert . Anschließend wurden Zellproben für weitere Experimente entnommen.RNA-Extraktion und -NachweisRNA wurde aus Lungengewebe und BMDMs mit dem RNA-Extraktionskit (R0026, Beyotime) isoliert. Die RNA-Konzentration wurde gemessen und verdünnt. Anschließend erfolgte die cDNA-Synthese mit dem SweScript RT cDNA-Synthesekit (G3333, Servicebio). Das TB Green PCR-Kit (G3320, Servicebio) wurde den Proben zugegeben. Die quantitative Echtzeit-PCR (qRT-PCR) wurde auf einem PCR-Gerät von Biosystems durchgeführt. Die Genexpression wurde im Vergleich zu den Kontrollen mit der 2<sup>-ΔΔCt</sup>-Formel analysiert. Die Expression wurde auf die mittlere Expression aller Kontrollprobanden normiert.Folgende Primer wurden verwendet: β-Actin, 5'-CACGATGGAGGGGCCGGACTCATC-3' (vorwärts) und 5' -TAAAGACCTCTATGCCAACACAGT-3' (rückwärts); NLRX1, 5'-CAGATTGGTAACAAAGGAGCCA-3' (vorwärts) und 5'-CGTTCGGTTTATCTTCAGAGCA-3' (rückwärts).Western BlotGesamtproteine wurden aus Lungengewebe und BMDMs isoliert und die Konzentration mittels BCA-Assay-Kit (P0012, Beyotime) quantifiziert. Proben mit 50 µg Protein wurden auf ein 10%iges SDS-PAGE-Gel aufgetragen und einer einstündigen Gelelektrophorese unterzogen. Anschließend wurden die Proteine auf PVDF-Membranen transferiert und mit Western-Blot-Blockierungslösung (P30500, NCM Biotech) behandelt. Die Membranen wurden über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-NLRX1 (17215-1-AP, 1:1000, Proteintech), anti-IL-1β (A16288, 1:1000, Abclonal), anti-IL-18 (A1115, 1:1000, Abclonal), anti-pNF-κB p65 (82335-1-RR, 1:1000, Proteintech), anti-NF-κB p65 (10745-1-AP, 1:1000, Proteintech), anti-Nrf2 (A0674, 1:1000, Abclonal), anti-Keap1 (10503-2-AP, 1:1000, Proteintech) und anti-β-Actin (ab8227, 1:5000, Abcam). Inkubation mit dem Sekundärantikörper (RGAR001, 1:5000, Proteintech) für 1 h. Der Nachweis erfolgte mittels Chemilumineszenzsystem (CLINX ChemiScope).ZytokinmessungNach Aspiration und Zentrifugation des Überstands aus der BMDM-Kultur wurde die Konzentration von IL-1β und IL-6 mittels ELISA-Kits (R&D) bestimmt. Die Gebrauchsanweisung wurde vom Hersteller bereitgestellt.Messung der Superoxiddismutase (SOD)-AktivitätDie Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die Gesamt-SOD-Aktivität im Überstand mithilfe des Gesamt-SOD-Assay-Kits (S0101S, Beyotime) gemäß den Herstellerangaben bestimmt.Messung der gesamten antioxidativen Kapazität (TAC)Die Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die antioxidative Gesamtkapazität (TAC) im Überstand mithilfe des Testkits zur Bestimmung der antioxidativen Gesamtkapazität (S0121, Beyotime) gemäß den Herstellerangaben bestimmt.Statistische AnalysenDie Ergebnisse wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt und mit GraphPad Prism 9.0 analysiert. Signifikante Unterschiede zwischen den beiden Gruppen wurden mittels t-Test nach Student ermittelt. Unterschiede zwischen mehreren Gruppen wurden durch eine einfaktorielle Varianzanalyse (ANOVA) mit gegebenenfalls anschließendem Bonferroni-Post-hoc-Test untersucht. Ein p-Wert < 0,05 wurde als statistisch signifikant angesehen.ErgebnisseHypoxie führte zu einer verminderten Expression von NLRX1 im Lungengewebe von Mäusen.NLRX1 ist für seine negative Regulation von Entzündungen und Oxidation bekannt. Der Einfluss von Hypoxie auf die NLRX1-Funktion wurde jedoch bisher nicht untersucht. Daher analysierten wir die NLRX1-Expression im Lungengewebe von Mäusen mit HPH. Unsere Ergebnisse (Abb.
1
A und zeigen eine Reduktion sowohl der mRNA- als auch der Proteinspiegel von NLRX1 im Lungengewebe von HPH-Mäusen im Vergleich zur Kontrollgruppe.Abb. 1
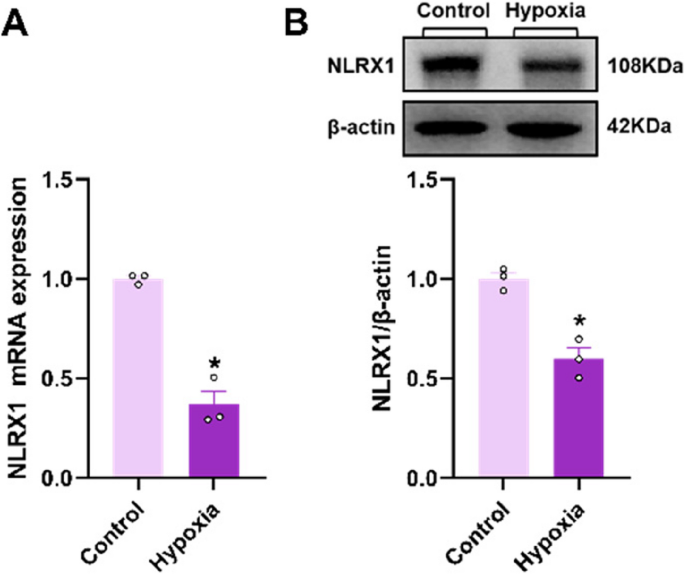 Die NLRX1-Expression ist im Lungengewebe von Mäusen mit Hypoxie verringert. A Die NLRX1-mRNA-Konzentration im Lungengewebe wurde mittels qPCR gemessen ( n = 3). B Die NLRX1-Protein-Expression wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle.
NLRX1-Mangel verschlimmerte die pulmonale Gefäßremodellierung bei HPH-Mäusen.In einer vorangegangenen Untersuchung beobachteten wir eine Herabregulierung von NLRX1 im Lungengewebe von HPH-Mäusen. Wir vermuteten daher, dass das Fehlen von NLRX1 die mit HPH assoziierte Gefäßremodellierung verstärken könnte. Um diese Vermutung zu überprüfen, wurden HPH-Mäuse (NLRX1<sup> −/−</sup>) anschließend einer Hypoxie ausgesetzt. Zur Beurteilung des Effekts der pulmonalen Gefäßremodellierung maßen wir die Dicke der Media der Pulmonalarterien. Die HE-Färbung zeigte einen deutlichen Anstieg des Wildtyp- (WT%) und des Weißmuskelanteils (WA%) in den Lungen von HPH-Mäusen im Vergleich zur Normoxie-Gruppe. Darüber hinaus waren die WT- und WA%-Werte von NLRX1<sup> −/− </sup>-Mäusen in der Hypoxie-Gruppe signifikant höher als die von Wildtyp-Mäusen. Die HE-Färbung zeigte außerdem, dass die Dicke der glatten Muskelschicht der Lunge bei NLRX1<sup> −/−</sup> -Mäusen in der Hypoxie-Gruppe diejenige von Wildtyp-Mäusen überstieg (Abb.
2
A und . Anschließend führten wir eine Immunfluoreszenzfärbung durch, um α-Aktin glatter Muskulatur (α-SMA) in Lungenschnitten zu markieren. Die Ergebnisse zeigten eine erhöhte Anzahl α-SMA-positiver Zellen, die die Lungengefäße in NLRX1<sup> −/−</sup> -Mäusen mit HPH im Vergleich zu Wildtyp-Mäusen umgeben, was die Verschlimmerung des Gefäßumbaus weiter belegt (Abb.
2C
).Abb. 2
Die NLRX1-Expression ist im Lungengewebe von Mäusen mit Hypoxie verringert. A Die NLRX1-mRNA-Konzentration im Lungengewebe wurde mittels qPCR gemessen ( n = 3). B Die NLRX1-Protein-Expression wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle.
NLRX1-Mangel verschlimmerte die pulmonale Gefäßremodellierung bei HPH-Mäusen.In einer vorangegangenen Untersuchung beobachteten wir eine Herabregulierung von NLRX1 im Lungengewebe von HPH-Mäusen. Wir vermuteten daher, dass das Fehlen von NLRX1 die mit HPH assoziierte Gefäßremodellierung verstärken könnte. Um diese Vermutung zu überprüfen, wurden HPH-Mäuse (NLRX1<sup> −/−</sup>) anschließend einer Hypoxie ausgesetzt. Zur Beurteilung des Effekts der pulmonalen Gefäßremodellierung maßen wir die Dicke der Media der Pulmonalarterien. Die HE-Färbung zeigte einen deutlichen Anstieg des Wildtyp- (WT%) und des Weißmuskelanteils (WA%) in den Lungen von HPH-Mäusen im Vergleich zur Normoxie-Gruppe. Darüber hinaus waren die WT- und WA%-Werte von NLRX1<sup> −/− </sup>-Mäusen in der Hypoxie-Gruppe signifikant höher als die von Wildtyp-Mäusen. Die HE-Färbung zeigte außerdem, dass die Dicke der glatten Muskelschicht der Lunge bei NLRX1<sup> −/−</sup> -Mäusen in der Hypoxie-Gruppe diejenige von Wildtyp-Mäusen überstieg (Abb.
2
A und . Anschließend führten wir eine Immunfluoreszenzfärbung durch, um α-Aktin glatter Muskulatur (α-SMA) in Lungenschnitten zu markieren. Die Ergebnisse zeigten eine erhöhte Anzahl α-SMA-positiver Zellen, die die Lungengefäße in NLRX1<sup> −/−</sup> -Mäusen mit HPH im Vergleich zu Wildtyp-Mäusen umgeben, was die Verschlimmerung des Gefäßumbaus weiter belegt (Abb.
2C
).Abb. 2
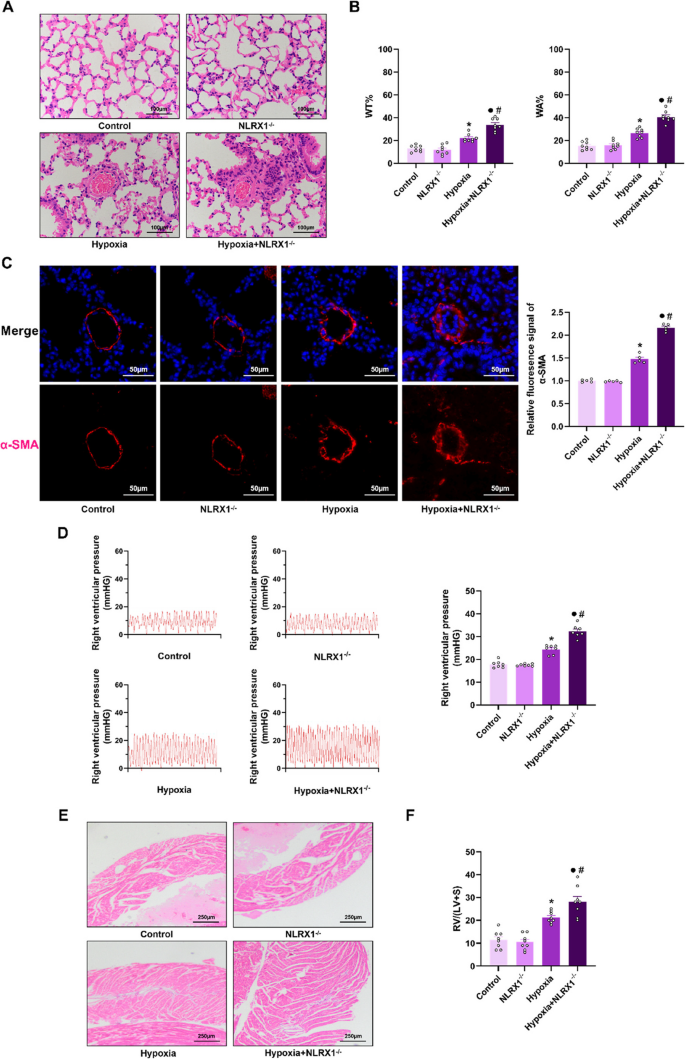 Der NLRX1-Knockout führt zu einer Verschlimmerung der hypoxieinduzierten pulmonalen Hypertonie. A Lungengewebe wurde nach Hämatoxylin-Eosin-Färbung histologisch mittels Lichtmikroskop analysiert. Maßstabsbalken: 100 μm. B WT% und WA% wurden zur Analyse der Lungenmorphometrie berechnet ( n = . C Immunfluoreszenzaufnahmen von Lungenschnitten, gefärbt für α-SMA (rot) und DAPI (blau), mit Quantifizierung ( n = 5). Maßstabsbalken: 50 μm. D Repräsentative Bilder der RVSP-Kurve mit Quantifizierung ( n = . E Herzgewebe wurde nach Hämatoxylin-Eosin-Färbung histologisch mittels Mikroskop analysiert. Maßstabsbalken: 250 μm. F RV/(LV + S) wurde zur Analyse der rechtsventrikulären Hypertrophie berechnet ( n = . Die Daten wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 −/− , # P < 0,05 vs. Hypoxie
NLRX1-vermittelte hämodynamische Reaktion und rechtsventrikuläre Hypertrophie bei HPH-MäusenHämodynamische Veränderungen, die primär auf vaskuläres Remodeling zurückzuführen sind, sind ein wichtiger Indikator für pulmonale Hypertonie. Die Gefäßdynamik wird durch die Messung des rechtsventrikulären systolischen Drucks (RVSP) beurteilt. Wie in Abb.
2D
dargestellt , führte die Exposition gegenüber Hypoxie zu einem signifikanten Anstieg des RVSP bei Mäusen mit pulmonaler Hypertonie (HPH). Insbesondere war der RVSP bei NLRX1<sup> −/−</sup> -Mäusen im Vergleich zu Wildtyp-Mäusen (WT) erhöht (Abb.
2D
). Diese hämodynamischen Veränderungen können den rechten Ventrikel zusätzlich belasten und letztendlich eine Rechtsherzhypertrophie verursachen. Die histologische Analyse zeigte eine Verdickung des rechten Ventrikels bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe, wobei die Hypertrophie bei NLRX1<sup> −/−</sup> -Mäusen stärker ausgeprägt war als bei WT-Mäusen (Abb.
2E
). Das Verhältnis RV/LV + S ist ein häufig verwendeter Indikator für die Rechtsherzhypertrophie. Die Ergebnisse zeigten einen Anstieg des RV/LV + S-Verhältnisses bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe. Unter hypoxischen Bedingungen wurden bei NLRX1<sup> −/−</sup> -Mäusen sogar noch höhere Werte beobachtet als bei Wildtyp-Mäusen (Abb.
2F
). Zusammenfassend deuten diese Befunde darauf hin, dass die Deletion von NLRX1 die HPH bei Mäusen verschlimmert.Ein NLRX1-Mangel führte bei HPH-Mäusen zu einer Hochregulierung von Entzündungsmediatoren durch Aktivierung von NF-κB im Lungengewebe.Hypoxie stimuliert bei pulmonaler Hypertonie primär Entzündungsreaktionen durch die Induktion der Produktion von Entzündungszytokinen wie IL-1β und IL-18. Um die Rolle von NLRX1 bei hypoxieinduzierter Entzündung zu untersuchen, bestimmten wir die Konzentrationen dieser Entzündungszytokine im Lungengewebe von Mäusen jeder Versuchsgruppe. Die IL-1β- und IL-18-Konzentrationen waren im Lungengewebe von Mäusen mit pulmonaler Hypertonie im Vergleich zur Normoxie-Gruppe erhöht. Darüber hinaus waren die Konzentrationen dieser Zytokine in NLRX1<sup> −/− </sup>-Mäusen mit pulmonaler Hypertonie signifikant höher als in Wildtyp-Mäusen (Abb.
3A
).Abb. 3
Der NLRX1-Knockout führt zu einer Verschlimmerung der hypoxieinduzierten pulmonalen Hypertonie. A Lungengewebe wurde nach Hämatoxylin-Eosin-Färbung histologisch mittels Lichtmikroskop analysiert. Maßstabsbalken: 100 μm. B WT% und WA% wurden zur Analyse der Lungenmorphometrie berechnet ( n = . C Immunfluoreszenzaufnahmen von Lungenschnitten, gefärbt für α-SMA (rot) und DAPI (blau), mit Quantifizierung ( n = 5). Maßstabsbalken: 50 μm. D Repräsentative Bilder der RVSP-Kurve mit Quantifizierung ( n = . E Herzgewebe wurde nach Hämatoxylin-Eosin-Färbung histologisch mittels Mikroskop analysiert. Maßstabsbalken: 250 μm. F RV/(LV + S) wurde zur Analyse der rechtsventrikulären Hypertrophie berechnet ( n = . Die Daten wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 −/− , # P < 0,05 vs. Hypoxie
NLRX1-vermittelte hämodynamische Reaktion und rechtsventrikuläre Hypertrophie bei HPH-MäusenHämodynamische Veränderungen, die primär auf vaskuläres Remodeling zurückzuführen sind, sind ein wichtiger Indikator für pulmonale Hypertonie. Die Gefäßdynamik wird durch die Messung des rechtsventrikulären systolischen Drucks (RVSP) beurteilt. Wie in Abb.
2D
dargestellt , führte die Exposition gegenüber Hypoxie zu einem signifikanten Anstieg des RVSP bei Mäusen mit pulmonaler Hypertonie (HPH). Insbesondere war der RVSP bei NLRX1<sup> −/−</sup> -Mäusen im Vergleich zu Wildtyp-Mäusen (WT) erhöht (Abb.
2D
). Diese hämodynamischen Veränderungen können den rechten Ventrikel zusätzlich belasten und letztendlich eine Rechtsherzhypertrophie verursachen. Die histologische Analyse zeigte eine Verdickung des rechten Ventrikels bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe, wobei die Hypertrophie bei NLRX1<sup> −/−</sup> -Mäusen stärker ausgeprägt war als bei WT-Mäusen (Abb.
2E
). Das Verhältnis RV/LV + S ist ein häufig verwendeter Indikator für die Rechtsherzhypertrophie. Die Ergebnisse zeigten einen Anstieg des RV/LV + S-Verhältnisses bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe. Unter hypoxischen Bedingungen wurden bei NLRX1<sup> −/−</sup> -Mäusen sogar noch höhere Werte beobachtet als bei Wildtyp-Mäusen (Abb.
2F
). Zusammenfassend deuten diese Befunde darauf hin, dass die Deletion von NLRX1 die HPH bei Mäusen verschlimmert.Ein NLRX1-Mangel führte bei HPH-Mäusen zu einer Hochregulierung von Entzündungsmediatoren durch Aktivierung von NF-κB im Lungengewebe.Hypoxie stimuliert bei pulmonaler Hypertonie primär Entzündungsreaktionen durch die Induktion der Produktion von Entzündungszytokinen wie IL-1β und IL-18. Um die Rolle von NLRX1 bei hypoxieinduzierter Entzündung zu untersuchen, bestimmten wir die Konzentrationen dieser Entzündungszytokine im Lungengewebe von Mäusen jeder Versuchsgruppe. Die IL-1β- und IL-18-Konzentrationen waren im Lungengewebe von Mäusen mit pulmonaler Hypertonie im Vergleich zur Normoxie-Gruppe erhöht. Darüber hinaus waren die Konzentrationen dieser Zytokine in NLRX1<sup> −/− </sup>-Mäusen mit pulmonaler Hypertonie signifikant höher als in Wildtyp-Mäusen (Abb.
3A
).Abb. 3
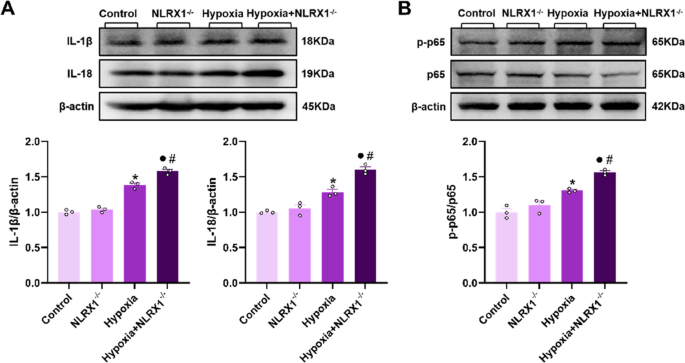 Der NLRX1-Knockout fördert die Aktivierung des NF-κB-Signalwegs und verschlimmert die Entzündung im Lungengewebe von PH-Mäusen. A Die Expression von IL-1β und IL-18 wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). B Die Phosphorylierung von NF-κB p65 wurde mittels Western Blot in Lungenlysaten gemessen und die Verhältnisse der molekularen Signalintensität quantifiziert. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
NF-κB ist ein wichtiger Transkriptionsfaktor, der die Bildung von Entzündungsmediatoren während der Entwicklung der pulmonalen Hypertonie (HPH) reguliert [
24
]. Aufgrund seiner vermittelnden Rolle untersuchten wir in unserer Studie den Einfluss von NLRX1 auf die NF-κB-Aktivierung im Lungengewebe von HPH-Mäusen. Unsere Ergebnisse zeigten, dass die Phosphorylierung von p65 im Lungengewebe von HPH-Mäusen im Vergleich zur Normoxie-Gruppe erhöht war. Besonders wichtig ist, dass das Fehlen von NLRX1 die p65-Phosphorylierung in der Hypoxie-Gruppe signifikant verstärkte (Abb.
3B
).Ein Mangel an NLRX1 förderte oxidativen Stress und die Aktivierung von Nrf2 im Lungengewebe von HPH-Mäusen.Bei hypoxämischer pulmonaler Hypertonie (HPH) führt eine durch Hypoxie bedingte mitochondriale Dysfunktion zu Störungen der Atmungskette und des Membranpotenzials, was die Produktion und Freisetzung reaktiver Sauerstoffspezies (ROS) zur Folge hat. Erhöhte ROS-Spiegel können verschiedene Signalwege aktivieren und so zu Entzündungen beitragen oder die Proliferation und Migration glatter Muskelzellen der Pulmonalarterie verstärken [
25
]. Obwohl bisherige Studien die mitochondriale Lokalisation von NLRX1 und dessen Rolle bei der Regulation der ROS-Produktion nachgewiesen haben, ist die spezifische Funktion von NLRX1 unter hypoxischen Bedingungen noch nicht vollständig geklärt. In dieser Studie untersuchten wir die Aktivitäten von TAC und SOD im Lungengewebe von Mäusen verschiedener Versuchsgruppen. Unsere Ergebnisse zeigten erhöhte TAC- und SOD-Aktivitäten im Lungengewebe von HPH-Mäusen im Vergleich zur Normoxie-Gruppe, wobei die Erhöhung bei NLRX1<sup> −/− </sup>-Mäusen unter hypoxischen Bedingungen deutlich stärker ausgeprägt war (Abb.
4
A und .Abb. 4
Der NLRX1-Knockout fördert die Aktivierung des NF-κB-Signalwegs und verschlimmert die Entzündung im Lungengewebe von PH-Mäusen. A Die Expression von IL-1β und IL-18 wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). B Die Phosphorylierung von NF-κB p65 wurde mittels Western Blot in Lungenlysaten gemessen und die Verhältnisse der molekularen Signalintensität quantifiziert. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
NF-κB ist ein wichtiger Transkriptionsfaktor, der die Bildung von Entzündungsmediatoren während der Entwicklung der pulmonalen Hypertonie (HPH) reguliert [
24
]. Aufgrund seiner vermittelnden Rolle untersuchten wir in unserer Studie den Einfluss von NLRX1 auf die NF-κB-Aktivierung im Lungengewebe von HPH-Mäusen. Unsere Ergebnisse zeigten, dass die Phosphorylierung von p65 im Lungengewebe von HPH-Mäusen im Vergleich zur Normoxie-Gruppe erhöht war. Besonders wichtig ist, dass das Fehlen von NLRX1 die p65-Phosphorylierung in der Hypoxie-Gruppe signifikant verstärkte (Abb.
3B
).Ein Mangel an NLRX1 förderte oxidativen Stress und die Aktivierung von Nrf2 im Lungengewebe von HPH-Mäusen.Bei hypoxämischer pulmonaler Hypertonie (HPH) führt eine durch Hypoxie bedingte mitochondriale Dysfunktion zu Störungen der Atmungskette und des Membranpotenzials, was die Produktion und Freisetzung reaktiver Sauerstoffspezies (ROS) zur Folge hat. Erhöhte ROS-Spiegel können verschiedene Signalwege aktivieren und so zu Entzündungen beitragen oder die Proliferation und Migration glatter Muskelzellen der Pulmonalarterie verstärken [
25
]. Obwohl bisherige Studien die mitochondriale Lokalisation von NLRX1 und dessen Rolle bei der Regulation der ROS-Produktion nachgewiesen haben, ist die spezifische Funktion von NLRX1 unter hypoxischen Bedingungen noch nicht vollständig geklärt. In dieser Studie untersuchten wir die Aktivitäten von TAC und SOD im Lungengewebe von Mäusen verschiedener Versuchsgruppen. Unsere Ergebnisse zeigten erhöhte TAC- und SOD-Aktivitäten im Lungengewebe von HPH-Mäusen im Vergleich zur Normoxie-Gruppe, wobei die Erhöhung bei NLRX1<sup> −/− </sup>-Mäusen unter hypoxischen Bedingungen deutlich stärker ausgeprägt war (Abb.
4
A und .Abb. 4
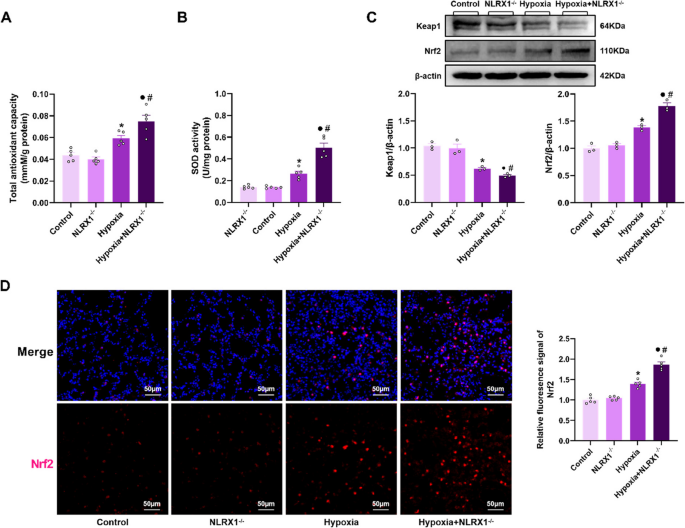 Das Fehlen von NLRX1 förderte die Hypoxie-induzierte Aktivierung des Keap1/Nrf2-Signalwegs im Lungengewebe von PH-Mäusen. A: Nachweis der totalen atmosphärischen Kapazität (TAC) im Lungengewebe von Mäusen ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) im Lungengewebe von Mäusen ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). D: Immunfluoreszenzaufnahmen von Lungenschnitten, gefärbt für Nrf2 (rot) und DAPI (blau), mit Quantifizierung ( n = 5). Maßstabsbalken: 50 μm. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
Unter oxidativem Stress können gebildete Oxidantien die Keap1-Nrf2-Interaktion stören, wodurch Nrf2 in den Zellkern transloziert und die Transkription antioxidativer Gene aktiviert wird [
26
]. Um den Einfluss von NLRX1 auf die Nrf2-Aktivierung bei HPH zu untersuchen, analysierten wir die Proteinexpressionsniveaus von Keap1 und Nrf2 im Lungengewebe von Mäusen. Die Ergebnisse zeigten verringerte Keap1-Spiegel und eine erhöhte Nrf2-Expression in den Lungen von HPH-Mäusen nach Hypoxieexposition. Der NLRX1-Mangel in HPH-Mäusen könnte diese Veränderung der Expressionsmuster verstärkt haben (Abb.
4C
). Darüber hinaus zeigte die Immunfluoreszenzfärbung von Nrf2 in Lungenschnitten eine signifikant höhere Anzahl Nrf2-positiver Zellen in NLRX1<sup> −/−</sup> -Mäusen im Vergleich zu Wildtyp-Mäusen. Dies deutet darauf hin, dass der NLRX1-Mangel die Bildung von Molekülen fördert, die oxidativen Stress induzieren, was zu einer übermäßigen Nrf2-Aktivierung führt (Abb.
4D
).Hypoxie beeinflusste die Rekrutierung von Immunzellen über NLRX1.Frühere Studien haben gezeigt, dass NLRX1 die Aktivierung von Makrophagen und T-Zellen hemmen kann. Um zu untersuchen, wie Hypoxie die Rekrutierung von Immunzellen über NLRX1 beeinflusst, wurde eine Immunfluoreszenzfärbung an CD4<sup> + </sup>-T-Zellen und Makrophagen in der Lunge von Mäusen durchgeführt. Die Ergebnisse in Abb.
5
zeigen, dass nach Hypoxiestimulation die Anzahl CD4-positiver Zellen in der Lunge von HPH-Mäusen signifikant abnahm, während die Anzahl F4/80-positiver Zellen deutlich zunahm. Darüber hinaus fanden sich in der Hypoxiegruppe mehr F4/80-positive Zellen in NLRX1<sup> −/−</sup> -Mäusen als in Wildtyp-Mäusen (Abb.
5B
), was darauf hindeutet, dass NLRX1 eher bei der Makrophagenrekrutierung als bei der Rekrutierung von CD4<sup> + </sup>-T-Zellen während HPH eine Rolle spielt.Abb. 5
Das Fehlen von NLRX1 förderte die Hypoxie-induzierte Aktivierung des Keap1/Nrf2-Signalwegs im Lungengewebe von PH-Mäusen. A: Nachweis der totalen atmosphärischen Kapazität (TAC) im Lungengewebe von Mäusen ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) im Lungengewebe von Mäusen ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in Lungenlysaten analysiert und die Signalintensität quantifiziert ( n = 3). D: Immunfluoreszenzaufnahmen von Lungenschnitten, gefärbt für Nrf2 (rot) und DAPI (blau), mit Quantifizierung ( n = 5). Maßstabsbalken: 50 μm. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
Unter oxidativem Stress können gebildete Oxidantien die Keap1-Nrf2-Interaktion stören, wodurch Nrf2 in den Zellkern transloziert und die Transkription antioxidativer Gene aktiviert wird [
26
]. Um den Einfluss von NLRX1 auf die Nrf2-Aktivierung bei HPH zu untersuchen, analysierten wir die Proteinexpressionsniveaus von Keap1 und Nrf2 im Lungengewebe von Mäusen. Die Ergebnisse zeigten verringerte Keap1-Spiegel und eine erhöhte Nrf2-Expression in den Lungen von HPH-Mäusen nach Hypoxieexposition. Der NLRX1-Mangel in HPH-Mäusen könnte diese Veränderung der Expressionsmuster verstärkt haben (Abb.
4C
). Darüber hinaus zeigte die Immunfluoreszenzfärbung von Nrf2 in Lungenschnitten eine signifikant höhere Anzahl Nrf2-positiver Zellen in NLRX1<sup> −/−</sup> -Mäusen im Vergleich zu Wildtyp-Mäusen. Dies deutet darauf hin, dass der NLRX1-Mangel die Bildung von Molekülen fördert, die oxidativen Stress induzieren, was zu einer übermäßigen Nrf2-Aktivierung führt (Abb.
4D
).Hypoxie beeinflusste die Rekrutierung von Immunzellen über NLRX1.Frühere Studien haben gezeigt, dass NLRX1 die Aktivierung von Makrophagen und T-Zellen hemmen kann. Um zu untersuchen, wie Hypoxie die Rekrutierung von Immunzellen über NLRX1 beeinflusst, wurde eine Immunfluoreszenzfärbung an CD4<sup> + </sup>-T-Zellen und Makrophagen in der Lunge von Mäusen durchgeführt. Die Ergebnisse in Abb.
5
zeigen, dass nach Hypoxiestimulation die Anzahl CD4-positiver Zellen in der Lunge von HPH-Mäusen signifikant abnahm, während die Anzahl F4/80-positiver Zellen deutlich zunahm. Darüber hinaus fanden sich in der Hypoxiegruppe mehr F4/80-positive Zellen in NLRX1<sup> −/−</sup> -Mäusen als in Wildtyp-Mäusen (Abb.
5B
), was darauf hindeutet, dass NLRX1 eher bei der Makrophagenrekrutierung als bei der Rekrutierung von CD4<sup> + </sup>-T-Zellen während HPH eine Rolle spielt.Abb. 5
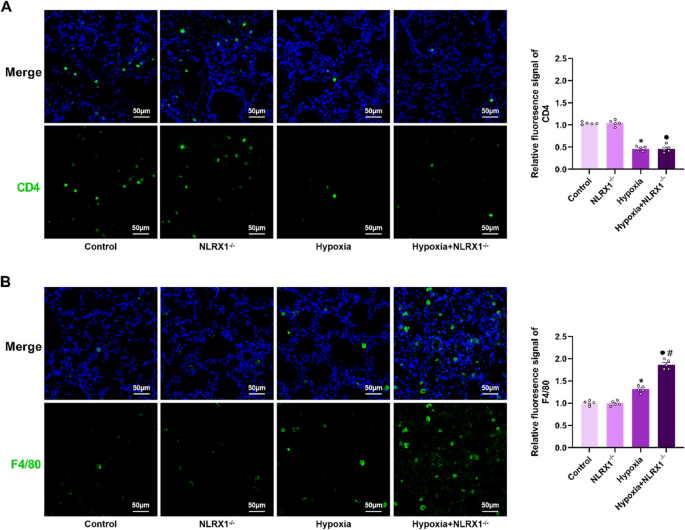 Hypoxie beeinflusst die Rekrutierung von Immunzellen in der Lunge von PH-Mäusen über NLRX1. Fluoreszenzgrüne Färbung von CD4 ( A ) und F4/80 ( B ). Die blaue Kernfärbung wurde mit DAPI-Färbelösung aktiviert (Maßstabsbalken: 50 μm). Quantifizierung ( n = 5). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
Hypoxie führte zu einer Herabregulierung der NLRX1-Expression in BMDMs.Unsere vorherige Untersuchung zeigte eine verminderte NLRX1-Expression im Lungengewebe bei HPH, begleitet von einer deutlichen Makrophagenakkumulation. Angesichts des engen Zusammenhangs zwischen Makrophagen, Entzündung und oxidativem Stress bei HPH ist es entscheidend, die entzündungshemmenden und antioxidativen Eigenschaften von NLRX1 in vivo zu identifizieren. Wir untersuchten die NLRX1-Expression in BMDMs. Nach Hypoxiestimulation sanken sowohl die mRNA- als auch die Proteinspiegel von NLRX1 in BMDMs nach 24 und 48 Stunden im Vergleich zur Kontrollgruppe (Abb.
6
A und .Abb. 6
Hypoxie beeinflusst die Rekrutierung von Immunzellen in der Lunge von PH-Mäusen über NLRX1. Fluoreszenzgrüne Färbung von CD4 ( A ) und F4/80 ( B ). Die blaue Kernfärbung wurde mit DAPI-Färbelösung aktiviert (Maßstabsbalken: 50 μm). Quantifizierung ( n = 5). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
Hypoxie führte zu einer Herabregulierung der NLRX1-Expression in BMDMs.Unsere vorherige Untersuchung zeigte eine verminderte NLRX1-Expression im Lungengewebe bei HPH, begleitet von einer deutlichen Makrophagenakkumulation. Angesichts des engen Zusammenhangs zwischen Makrophagen, Entzündung und oxidativem Stress bei HPH ist es entscheidend, die entzündungshemmenden und antioxidativen Eigenschaften von NLRX1 in vivo zu identifizieren. Wir untersuchten die NLRX1-Expression in BMDMs. Nach Hypoxiestimulation sanken sowohl die mRNA- als auch die Proteinspiegel von NLRX1 in BMDMs nach 24 und 48 Stunden im Vergleich zur Kontrollgruppe (Abb.
6
A und .Abb. 6
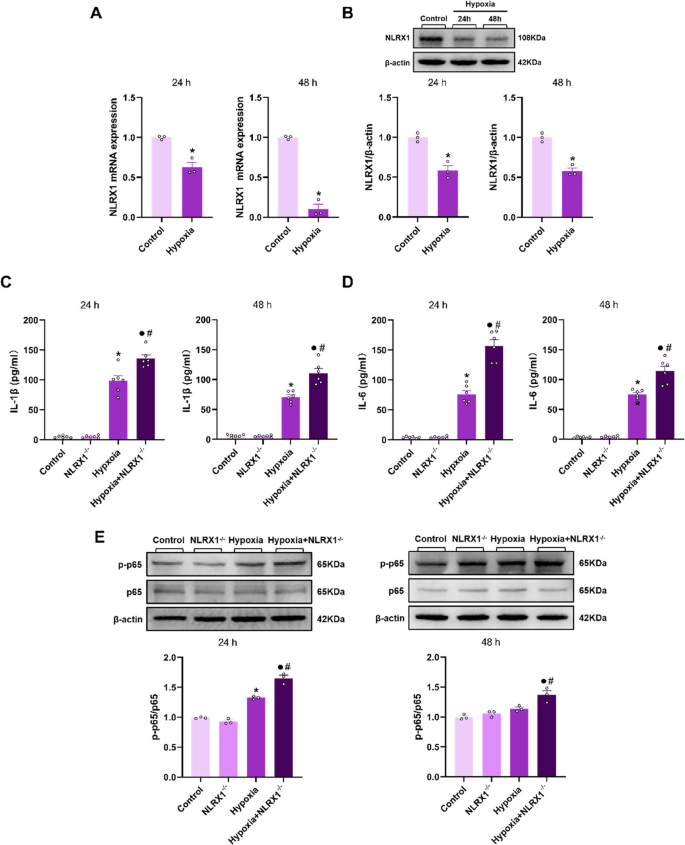 Die NLRX1-Expression ist in hypoxieinduzierten BMDMs verringert, und ein NLRX1-Mangel führt zu einer Überexpression von NF-κB und fördert die Produktion von Entzündungsfaktoren. A Die NLRX1-mRNA-Spiegel wurden mittels Real-Time-PCR gemessen ( n = 3). B Die NLRX1-Protein-Expression wurde mittels Western Blot unter Verwendung von BMDM-Lysaten analysiert und die Signalintensität von NLRX1 quantifiziert ( n = 3). C und D Die Konzentrationen von IL-1β und IL-6 im BMDM-Kulturmedium wurden mittels ELISA gemessen ( n = 6). E Die Phosphorylierung von NF-κB p65 wurde mittels Western Blot unter Verwendung von BMDM-Lysaten gemessen und die Verhältnisse der molekularen Signalintensität quantifiziert. Die Daten wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 −/− , # P < 0,05 vs. Hypoxie
NLRX1-Mangel führte zu einer Hochregulierung der Produktion entzündungsfördernder Zytokine durch Förderung der NF-κB-Aktivierung in hypoxiestimulierten BMDMs.Die Konzentration entzündungsfördernder Zytokine im Kulturmedium von BMDMs verschiedener Gruppen wurde gemessen. Nach 24 und 48 Stunden Hypoxiestimulation zeigten sowohl WT- als auch NLRX1<sup> −/−</sup> -BMDMs einen Anstieg der IL-1β- und IL-6-Spiegel im Kulturmedium. Bemerkenswerterweise wiesen NLRX1<sup> −/−</sup> -BMDMs in der Hypoxiegruppe zu jedem Messzeitpunkt höhere Zytokinkonzentrationen auf als WT-BMDMs (Abb.
6
C und D).Wir hatten zuvor festgestellt, dass NF-κB im Lungengewebe von NLRX1<sup> −/−</sup> -Mäusen mit HPH überaktiviert ist. Zusätzlich zu dieser Entdeckung untersuchten wir die NF-κB-Aktivierung in BMDMs. Dabei stellten wir fest, dass die Phosphorylierung von p65 in WT-BMDMs nach Hypoxie-Stimulation nach 24 Stunden im Vergleich zur Normoxie-Gruppe erhöht war. Darüber hinaus war die Phosphorylierung von p65 in NLRX1<sup> −/−</sup> -BMDMs der Hypoxie-Gruppe zu jedem Messzeitpunkt sowohl im Vergleich zu WT-BMDMs als auch zur Normoxie-Gruppe signifikant erhöht (Abb.
6E
).NLRX1-Mangel förderte oxidativen Stress und die Aktivierung von Nrf2 in hypoxiestimulierten BMDMs.Es wurde festgestellt, dass das Fehlen von NLRX1 zur Aktivierung von Nrf2 und einer erhöhten antioxidativen Kapazität im Lungengewebe von HPH-Mäusen beiträgt. Um die potenzielle Rolle von NLRX1 als Antioxidans in Makrophagen zu untersuchen, bestimmten wir die TAC- und SOD-Aktivität in BMDMs verschiedener Versuchsgruppen. Nach Hypoxie-Stimulation stieg die TAC- und SOD-Aktivität in BMDMs nach 24 h an, zeigte aber nach 48 h im Vergleich zur Normoxie-Gruppe keinen signifikanten Unterschied. Bemerkenswerterweise wiesen NLRX1<sup> −/−</sup> -BMDMs nach 24 h Hypoxie-Stimulation signifikant höhere TAC- und SOD-Aktivitäten auf als WT-BMDMs (Abb.
7
A und . Die anschließende Analyse der Keap1- und Nrf2-Expression in BMDMs ergab, dass die Keap1-Expression in WT-BMDMs nach 24 h Hypoxie abnahm, während die Nrf2-Expression im Vergleich zur Normoxie-Gruppe anstieg, ohne dass nach 48 h signifikante Unterschiede festgestellt wurden. Wichtig ist, dass die Deletion von NLRX1 zu jedem Zeitpunkt im Vergleich zu WT-BMDMs und der Normoxie-Gruppe deutlichere Veränderungen der Keap1- und Nrf2-Expression in hypoxieinduzierten BMDMs hervorrief (Abb.
7
C).Abb. 7
Die NLRX1-Expression ist in hypoxieinduzierten BMDMs verringert, und ein NLRX1-Mangel führt zu einer Überexpression von NF-κB und fördert die Produktion von Entzündungsfaktoren. A Die NLRX1-mRNA-Spiegel wurden mittels Real-Time-PCR gemessen ( n = 3). B Die NLRX1-Protein-Expression wurde mittels Western Blot unter Verwendung von BMDM-Lysaten analysiert und die Signalintensität von NLRX1 quantifiziert ( n = 3). C und D Die Konzentrationen von IL-1β und IL-6 im BMDM-Kulturmedium wurden mittels ELISA gemessen ( n = 6). E Die Phosphorylierung von NF-κB p65 wurde mittels Western Blot unter Verwendung von BMDM-Lysaten gemessen und die Verhältnisse der molekularen Signalintensität quantifiziert. Die Daten wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 −/− , # P < 0,05 vs. Hypoxie
NLRX1-Mangel führte zu einer Hochregulierung der Produktion entzündungsfördernder Zytokine durch Förderung der NF-κB-Aktivierung in hypoxiestimulierten BMDMs.Die Konzentration entzündungsfördernder Zytokine im Kulturmedium von BMDMs verschiedener Gruppen wurde gemessen. Nach 24 und 48 Stunden Hypoxiestimulation zeigten sowohl WT- als auch NLRX1<sup> −/−</sup> -BMDMs einen Anstieg der IL-1β- und IL-6-Spiegel im Kulturmedium. Bemerkenswerterweise wiesen NLRX1<sup> −/−</sup> -BMDMs in der Hypoxiegruppe zu jedem Messzeitpunkt höhere Zytokinkonzentrationen auf als WT-BMDMs (Abb.
6
C und D).Wir hatten zuvor festgestellt, dass NF-κB im Lungengewebe von NLRX1<sup> −/−</sup> -Mäusen mit HPH überaktiviert ist. Zusätzlich zu dieser Entdeckung untersuchten wir die NF-κB-Aktivierung in BMDMs. Dabei stellten wir fest, dass die Phosphorylierung von p65 in WT-BMDMs nach Hypoxie-Stimulation nach 24 Stunden im Vergleich zur Normoxie-Gruppe erhöht war. Darüber hinaus war die Phosphorylierung von p65 in NLRX1<sup> −/−</sup> -BMDMs der Hypoxie-Gruppe zu jedem Messzeitpunkt sowohl im Vergleich zu WT-BMDMs als auch zur Normoxie-Gruppe signifikant erhöht (Abb.
6E
).NLRX1-Mangel förderte oxidativen Stress und die Aktivierung von Nrf2 in hypoxiestimulierten BMDMs.Es wurde festgestellt, dass das Fehlen von NLRX1 zur Aktivierung von Nrf2 und einer erhöhten antioxidativen Kapazität im Lungengewebe von HPH-Mäusen beiträgt. Um die potenzielle Rolle von NLRX1 als Antioxidans in Makrophagen zu untersuchen, bestimmten wir die TAC- und SOD-Aktivität in BMDMs verschiedener Versuchsgruppen. Nach Hypoxie-Stimulation stieg die TAC- und SOD-Aktivität in BMDMs nach 24 h an, zeigte aber nach 48 h im Vergleich zur Normoxie-Gruppe keinen signifikanten Unterschied. Bemerkenswerterweise wiesen NLRX1<sup> −/−</sup> -BMDMs nach 24 h Hypoxie-Stimulation signifikant höhere TAC- und SOD-Aktivitäten auf als WT-BMDMs (Abb.
7
A und . Die anschließende Analyse der Keap1- und Nrf2-Expression in BMDMs ergab, dass die Keap1-Expression in WT-BMDMs nach 24 h Hypoxie abnahm, während die Nrf2-Expression im Vergleich zur Normoxie-Gruppe anstieg, ohne dass nach 48 h signifikante Unterschiede festgestellt wurden. Wichtig ist, dass die Deletion von NLRX1 zu jedem Zeitpunkt im Vergleich zu WT-BMDMs und der Normoxie-Gruppe deutlichere Veränderungen der Keap1- und Nrf2-Expression in hypoxieinduzierten BMDMs hervorrief (Abb.
7
C).Abb. 7
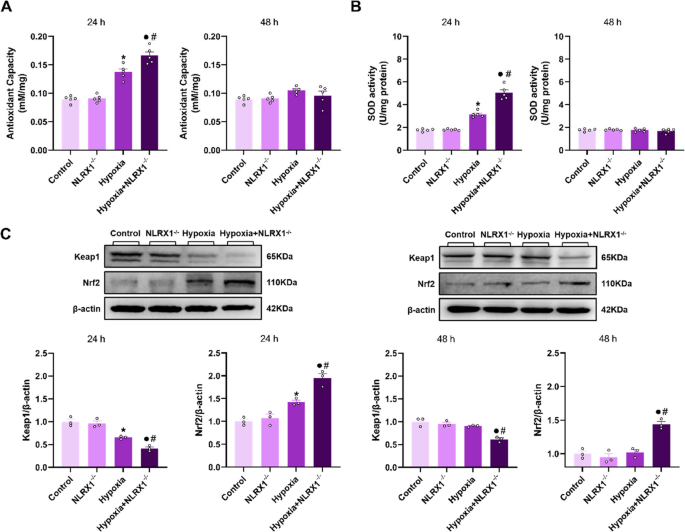 Das Fehlen von NLRX1 fördert die Aktivierung des Keap1/Nrf2-Signalwegs in hypoxieinduzierten BMDMs und erhöht die antioxidative Kapazität. A: Nachweis der Gesamtantioxidationskapazität (TAC) in BMDMs ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) in BMDMs ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in BMDM-Lysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
NX-13 reduzierte die Produktion entzündungsfördernder Zytokine durch Abschwächung der NF-κB-Aktivierung in hypoxiestimulierten BMDMs.NX-13, eine symmetrische Triphenylverbindung, wurde gezielt entwickelt, um NLRX1 zu aktivieren und dadurch entzündungshemmende und antioxidative Eigenschaften zu entfalten. Jüngste Forschungsergebnisse zeigen, dass NX-13 die Differenzierung und den Metabolismus von T-Zellen über NF-κB modulieren kann [
8
]. Um zu untersuchen, wie NX-13 die Makrophagenfunktion unter hypoxischen Bedingungen beeinflusst, bestimmten wir die Konzentrationen entzündungsfördernder Zytokine im Medium von BMDMs nach Vorbehandlung mit NX-13. Unsere Ergebnisse zeigen, dass die NX-13-Vorbehandlung die Konzentration von IL-1β und IL-6 im BMDM-Medium 24 und 48 Stunden nach Hypoxieinduktion effektiv reduziert (Abb.
8
A und . Darüber hinaus untersuchten wir die Aktivierung von NF-κB in BMDMs und beobachteten eine verminderte Phosphorylierung von p65 in hypoxieinduzierten BMDMs zu jedem Zeitpunkt nach NX-13-Vorbehandlung (Abb.
8
C).Abb. 8
Das Fehlen von NLRX1 fördert die Aktivierung des Keap1/Nrf2-Signalwegs in hypoxieinduzierten BMDMs und erhöht die antioxidative Kapazität. A: Nachweis der Gesamtantioxidationskapazität (TAC) in BMDMs ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) in BMDMs ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in BMDM-Lysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 <sup>−/−</sup> , # P < 0,05 vs. Hypoxie.
NX-13 reduzierte die Produktion entzündungsfördernder Zytokine durch Abschwächung der NF-κB-Aktivierung in hypoxiestimulierten BMDMs.NX-13, eine symmetrische Triphenylverbindung, wurde gezielt entwickelt, um NLRX1 zu aktivieren und dadurch entzündungshemmende und antioxidative Eigenschaften zu entfalten. Jüngste Forschungsergebnisse zeigen, dass NX-13 die Differenzierung und den Metabolismus von T-Zellen über NF-κB modulieren kann [
8
]. Um zu untersuchen, wie NX-13 die Makrophagenfunktion unter hypoxischen Bedingungen beeinflusst, bestimmten wir die Konzentrationen entzündungsfördernder Zytokine im Medium von BMDMs nach Vorbehandlung mit NX-13. Unsere Ergebnisse zeigen, dass die NX-13-Vorbehandlung die Konzentration von IL-1β und IL-6 im BMDM-Medium 24 und 48 Stunden nach Hypoxieinduktion effektiv reduziert (Abb.
8
A und . Darüber hinaus untersuchten wir die Aktivierung von NF-κB in BMDMs und beobachteten eine verminderte Phosphorylierung von p65 in hypoxieinduzierten BMDMs zu jedem Zeitpunkt nach NX-13-Vorbehandlung (Abb.
8
C).Abb. 8
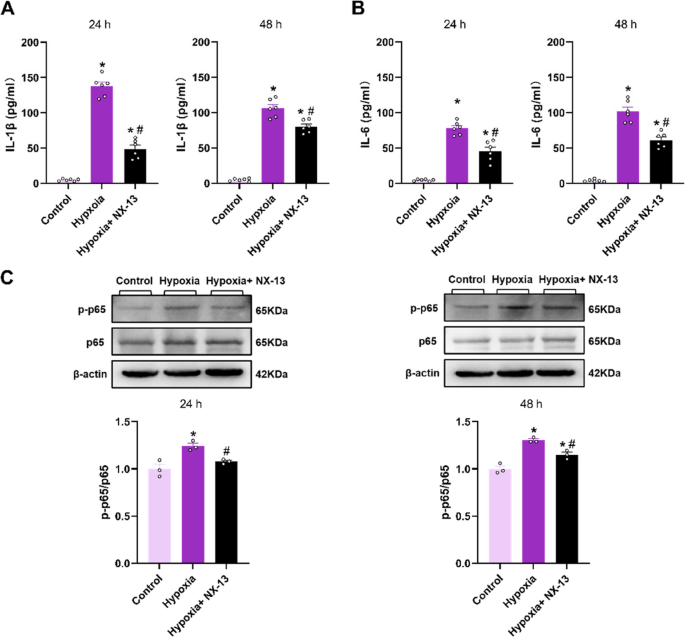 NX-13 hemmt die Aktivierung des NF-κB-Signalwegs in hypoxieinduzierten BMDMs und reduziert die Produktion von Entzündungsfaktoren. A und B: Die Konzentrationen von IL-1β und IL-6 im Kulturmedium von BMDMs wurden mittels ELISA bestimmt ( n = 6). C: Die Phosphorylierung von NF-κB p65 wurde mittels Western-Blot-Analyse unter Verwendung von BMDM-Lysaten gemessen und die Verhältnisse der molekularen Signalintensitäten quantifiziert. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle, # p < 0,05 vs. Hypoxie.
NX-13 linderte oxidativen Stress und schwächte die Nrf2-Aktivierung in hypoxiestimulierten BMDMs ab.Es wurde festgestellt, dass NX-13 die Expression antioxidativer Enzyme erhöht und die ROS-Bildung in T-Zellen hemmt. Daher untersuchten wir die TAC- und SOD-Aktivitäten in BMDMs. Bemerkenswert ist, dass die Vorbehandlung mit NX-13 nach 24 h im Vergleich zur Hypoxiegruppe zu einer Reduktion der TAC- und SOD-Aktivitäten führte, während nach 48 h kein signifikanter Unterschied mehr beobachtet wurde (Abb.
9
A und . Frühere Untersuchungen unserer Gruppe zeigten, dass das Fehlen von NLRX1 die Nrf2-Aktivierung in Lungengewebe und BMDMs von HPH-Mäusen verstärkt. Anschließend bestimmten wir die Keap1- und Nrf2-Spiegel in BMDMs. Interessanterweise stellte die NX-13-Vorbehandlung die Keap1-Expression nach 24 h Hypoxieexposition wieder her, reduzierte jedoch die Nrf2-Expression zu jedem Messzeitpunkt (Abb.
9
C). Aus den Ergebnissen von Tier- und Zellstudien schließen wir, dass NX-13 oxidativen Stress und die NF-κB-Aktivierung durch die Stimulation von NLRX1 in Makrophagen abschwächen und dadurch die Zellaggregation verringern sowie die Produktion von entzündungsfördernden Zytokinen reduzieren kann.Abb. 9
NX-13 hemmt die Aktivierung des NF-κB-Signalwegs in hypoxieinduzierten BMDMs und reduziert die Produktion von Entzündungsfaktoren. A und B: Die Konzentrationen von IL-1β und IL-6 im Kulturmedium von BMDMs wurden mittels ELISA bestimmt ( n = 6). C: Die Phosphorylierung von NF-κB p65 wurde mittels Western-Blot-Analyse unter Verwendung von BMDM-Lysaten gemessen und die Verhältnisse der molekularen Signalintensitäten quantifiziert. Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle, # p < 0,05 vs. Hypoxie.
NX-13 linderte oxidativen Stress und schwächte die Nrf2-Aktivierung in hypoxiestimulierten BMDMs ab.Es wurde festgestellt, dass NX-13 die Expression antioxidativer Enzyme erhöht und die ROS-Bildung in T-Zellen hemmt. Daher untersuchten wir die TAC- und SOD-Aktivitäten in BMDMs. Bemerkenswert ist, dass die Vorbehandlung mit NX-13 nach 24 h im Vergleich zur Hypoxiegruppe zu einer Reduktion der TAC- und SOD-Aktivitäten führte, während nach 48 h kein signifikanter Unterschied mehr beobachtet wurde (Abb.
9
A und . Frühere Untersuchungen unserer Gruppe zeigten, dass das Fehlen von NLRX1 die Nrf2-Aktivierung in Lungengewebe und BMDMs von HPH-Mäusen verstärkt. Anschließend bestimmten wir die Keap1- und Nrf2-Spiegel in BMDMs. Interessanterweise stellte die NX-13-Vorbehandlung die Keap1-Expression nach 24 h Hypoxieexposition wieder her, reduzierte jedoch die Nrf2-Expression zu jedem Messzeitpunkt (Abb.
9
C). Aus den Ergebnissen von Tier- und Zellstudien schließen wir, dass NX-13 oxidativen Stress und die NF-κB-Aktivierung durch die Stimulation von NLRX1 in Makrophagen abschwächen und dadurch die Zellaggregation verringern sowie die Produktion von entzündungsfördernden Zytokinen reduzieren kann.Abb. 9
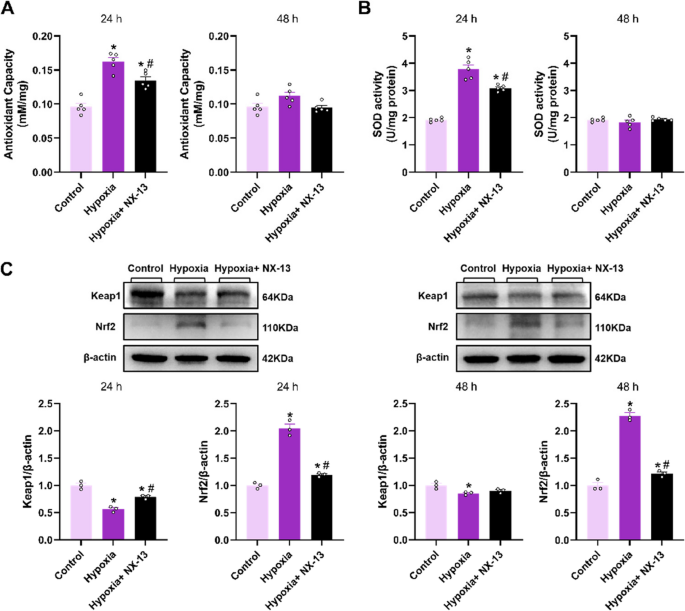 NX-13 hemmt die Aktivierung des Keap1/Nrf2-Signalwegs und beeinträchtigt die antioxidative Kapazität hypoxieinduzierter BMDMs. A: Nachweis der totalen antioxidativen Kapazität (TAC) in BMDMs ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) in BMDMs ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in BMDM-Lysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle, # p < 0,05 vs. Hypoxie.
DiskussionUnsere Studie zeigt, dass Hypoxie die NLRX1-Expression im Lungengewebe und in Makrophagen von Mäusen herabreguliert. Wir liefern erstmals den Beweis, dass ein NLRX1-Mangel bei HPH-Mäusen zu einem signifikanten Umbau der Pulmonalarterien und einer Hypertrophie des rechten Ventrikels führt, begleitet von einem deutlichen Anstieg der Lungenmakrophagen und entzündungsfördernder Zytokine. Diese Ergebnisse legen nahe, dass die Schutzfunktion von NLRX1 bei HPH die Suppression der Makrophagenaktivität und -infiltration, möglicherweise durch negative Regulation von oxidativem Stress und NF-κB-Aktivierung, umfassen könnte.Pathologisch ist die HPH durch einen erhöhten pulmonalarteriellen Druck gekennzeichnet, der zu einer Hypertrophie des rechten Ventrikels führt. Die vaskuläre Remodellierung der Pulmonalarterie ist ein wichtiger auslösender Faktor in diesem Prozess [
27
]. Hypoxie induziert eine abnorme Proliferation und Migration von PASMCs, was zu einer fortschreitenden Lumeneinengung, einer verminderten Gefäßcompliance, einem erhöhten Gefäßwiderstand und der Entwicklung einer pulmonalen Hypertonie führt [
28
]. Gleichzeitig passt sich der rechte Ventrikel dem erhöhten pulmonalen Druck durch eine Steigerung der myokardialen Kontraktilität an, was zu einer Kardiomyozytenhypertrophie führt [
29
]. NLRX1, ein Mitglied der NLR-Familie, fungiert als Mustererkennungsrezeptor. Jüngste Erkenntnisse deuten darauf hin, dass NLRX1 eine negative regulatorische Rolle bei verschiedenen Infektions- und Autoimmunerkrankungen spielt. Eine Dysregulation der NLRX1-Expression oder -Aktivität verschlimmert häufig die Krankheitsprogression [
30
,
31
].In dieser Studie simulierten wir mithilfe einer Niederdruck-Sauerstoffkammer die hypoxischen Bedingungen in 5500 m Höhe und induzierten so eine pulmonale Hypertonie bei Mäusen. Nach der hypoxieinduzierten pulmonalen Hypertonie wurde in den Lungengeweben von Wildtyp-Mäusen (WT) eine verminderte NLRX1-Expression beobachtet. Histopathologische Untersuchungen zeigten, dass der Mangel an NLRX1 zu ausgeprägten pathologischen Veränderungen in Lunge und rechtem Ventrikel der Mäuse mit hypoxämischer pulmonaler Hypertonie (HPH) führte. Ein Vergleich zwischen WT- und NLRX1 <sup>−/−</sup> -Mäusen nach HPH-Induktion ergab ein erhöhtes Verhältnis von WT- zu WA-Prozentwerten in NLRX1<sup> −/−</sup> -Mäusen, was auf eine abnorme Verengung des pulmonalen Gefäßlumens hindeutet. Zusätzlich deutete ein deutlicher Anstieg der α-SMA-positiven Zellen auf eine abnorme Proliferation glatter Muskelzellen hin. Darüber hinaus wiesen NLRX1 -Knockout- Mäuse im Vergleich zu Wildtyp-Mäusen einen höheren rechtsventrikulären systolischen Druck (RVSP) und ein höheres Verhältnis von rechtsventrikulärem zu linksventrikulärem und systolischem Druck (RV/(LV + S)) auf. Zusammengenommen deuten unsere Ergebnisse auf eine mögliche Schutzfunktion von NLRX1 bei HPH hin. Pathologisch betrachtet verschlimmert das Fehlen von NLRX1 das Remodeling der Pulmonalarterie und die rechtsventrikuläre Hypertrophie, was zu einem erhöhten pulmonalarteriellen Druck führt.Längerer Sauerstoffmangel induziert oxidativen Stress und Entzündungen, Schlüsselfaktoren in der Entwicklung der pulmonalen Hypertonie, indem er die Proliferation und Migration von PASMCs und das nachfolgende Gefäßremodeling verstärkt. Sauerstoffmangel stört die Funktion der mitochondrialen Atmungskette und führt zur Bildung reaktiver Sauerstoffspezies (ROS) [
32
]. ROS können IKKα direkt oxidieren oder über TLR4 den NF-κB-Signalweg aktivieren und so eine anhaltende Entzündungsreaktion auslösen [
33
,
34
]. Die Entzündung wird durch NLRs moduliert, welche Multiproteinkomplexe bilden und Signalwege aktivieren, die in der Sekretion proinflammatorischer Zytokine wie IL-1β, IL-6 und IL-18 münden [
35
]. Studien legen nahe, dass NLRX1 zelluläre Prozesse indirekt moduliert, indem es den ROS-Spiegel kontrolliert und die TLR4-vermittelte NF-κB-Aktivierung durch Interaktion mit TRAF6 in aktivierten Immunzellen hemmt [
36
]. Dennoch bleibt der genaue Regulationsmechanismus von NLRX1 auf NF-κB unter hypoxischen Bedingungen unklar.Wir untersuchten den oxidativen Stress in der Lunge von Wildtyp- (WT) und NLRX1 - Knockout-Mäusen (NLRX1 −/−) durch Analyse der NF-κB-Aktivierung und der Konzentrationen entzündungsfördernder Zytokine. Die Analyse zeigte, dass NLRX1 −/−- Mäuse nach HPH im Vergleich zu WT-Mäusen signifikant erhöhte TAC- und SOD-Aktivitäten im Lungengewebe aufwiesen, begleitet von einer gesteigerten Nrf2-Aktivierung. Dies deutet auf eine erhöhte Produktion von Substanzen des oxidativen Stresses, einschließlich reaktiver Sauerstoffspezies (ROS), hin. Darüber hinaus waren die NF-κB-Signalaktivierung sowie die Produktion der Zytokine IL-1β und IL-6 im Lungengewebe von NLRX1 −/−- Mäusen deutlich höher als in dem von WT-Mäusen. Diese Ergebnisse legen eine zentrale Rolle von übermäßigem oxidativem Stress und Entzündungsreaktionen in der Pathogenese der schweren HPH mit NLRX1-Deletion nahe. Obwohl Nrf2- und NF-κB-Signalwege im Lungengewebe von NLRX1<sup> −/−</sup> -Mäusen während der hypoxisch-pulmonalen Hypertonie (HPH) gleichzeitig aktiviert waren, zeigte sich keine offensichtliche Korrelation zwischen ihnen. Wir berichteten bereits über einen signifikanten Unterschied in der Anzahl der Makrophagen im Lungengewebe von HPH-Mäusen zwischen Wildtyp- und NLRX1<sup> −/−</sup> -Gruppen. Es ist jedoch weiterhin unklar, ob NLRX1 die Hyperaktivierung von Makrophagen über einen ähnlichen Mechanismus fördert. Um diese Frage zu klären, führten wir eine Ko-Färbung von NLRX1 und F4/80 im Lungengewebe von Mäusen durch und stellten fest, dass Hypoxie die Expression von NLRX1 in pulmonalen Makrophagen herunterreguliert (Abbildung
S1
). Anschließend verwendeten wir primäre Knochenmark-abgeleitete Makrophagen (BMDMs) von NLRX1<sup> −/−</sup> -Mäusen und unsere In-vitro-Untersuchungen zeigten, dass NLRX1 in hypoxieinduzierten BMDMs herunterreguliert war. Zusammenfassend deuten unsere Ergebnisse darauf hin, dass Hypoxie oxidativen Stress auslöst und NF-κB/Nrf2 in Lungengewebe und Makrophagen aktiviert. Diese Reaktion wird durch einen NLRX1-Mangel deutlich verstärkt. Diese Verstärkung korreliert mit einer erhöhten Produktion von Entzündungszytokinen in vivo und in vitro. Die Ergebnisse legen nahe, dass die Aktivierung oder Hochregulierung von NLRX1 eine übermäßige Immunaktivierung in Makrophagen durch Modulation des oxidativen Stresses abschwächen könnte. Um die Schutzwirkung von NLRX1 unter hypoxischen Bedingungen weiter zu untersuchen, behandelten wir BMDMs mit NX-13, einem NLRX1-Agonisten, vor. Die Vorbehandlung mit NX-13 unterdrückte nicht nur den hypoxieinduzierten oxidativen Stress und die NF-κB-Aktivierung in BMDMs, sondern verringerte auch die Produktion von Entzündungszytokinen und die Nrf2-Aktivierung.NLRX1 moduliert bemerkenswerterweise auch andere wichtige Signalwege der pulmonalen Hypertonie (PH). Ein Mangel an NLRX1 verstärkt die NF-κB-Aktivierung und erhöht die Konzentration proinflammatorischer Zytokine in HPH-Mäusen, was mit seiner Rolle bei der Hemmung der NF-κB- und NLRP3-Inflammasom-Aktivierung übereinstimmt. Präklinische Studien haben gezeigt, dass NLRX1-KO-Mäuse unter Stress eine verstärkte kardiale Entzündung und NLRP3-Aktivierung aufweisen [
37
], was mit der Beobachtung übereinstimmt, dass ein NLRX1-Mangel die pulmonale Entzündung bei HPH verschlimmert. NLRX1 reguliert zudem die ROS-Produktion und die FUNDC1-vermittelte Mitophagie [
38
]. HPH ist durch übermäßige mitochondriale Fusion, gestörte Mitophagie und oxidativen Stress gekennzeichnet. Unsere Studie zeigt, dass ein Mangel an NLRX1 die ROS-Produktion, die Gesamt-Antioxidationskapazität (TAC), die SOD-Aktivität und die Nrf2-Aktivierung erhöht, während sein Verlust mitochondriale Defekte in Kardiomyozyten verschlimmert. Dies legt nahe, dass NLRX1 die pulmonale Hypertonie (HPH) durch mitochondriale Regulation verhindern kann. Indirekt zeigt die druckinduzierte Herzhypertrophie eine Herunterregulierung von NLRX1, und seine Überexpression stellt die mitochondriale Funktion wieder her, was den Befunden bei HPH entspricht [
39
]. Darüber hinaus zeigt der Human Protein Atlas, dass NLRX1 in humanen Lungenmakrophagen vorkommt, was mit unseren Beobachtungen an Mäusen übereinstimmt und die Relevanz für die menschliche Erkrankung unterstreicht [
40
]. Zusammenfassend zeigt unsere Studie, dass Hypoxie die NLRX1-Expression reduziert, was zu einer verstärkten Makrophageninfiltration, erhöhtem oxidativem Stress und einer Hyperaktivierung von NF-κB führt. Frühere Untersuchungen deuten darauf hin, dass NLRX1 die ROS-Produktion bei akuten Infektionen stimulieren und dadurch die angeborene Immunantwort verstärken kann [
41
,
42
,
43
]. Längerer Sauerstoffmangel führt jedoch zu anhaltendem oxidativem Stress und erhöhten Konzentrationen proinflammatorischer Mediatoren, was letztendlich zu chronischer Entzündung und pulmonaler Hypertonie führt (Abb.
10
). Daher fungiert NLRX1 wahrscheinlich als negativer Regulator der ROS-Bildung bei chronischer Entzündung. Um die Rolle von NLRX1 bei HPH zu klären, sind weitere Untersuchungen zur Aufklärung der zugrundeliegenden Mechanismen erforderlich.Abb. 10
NX-13 hemmt die Aktivierung des Keap1/Nrf2-Signalwegs und beeinträchtigt die antioxidative Kapazität hypoxieinduzierter BMDMs. A: Nachweis der totalen antioxidativen Kapazität (TAC) in BMDMs ( n = 5). B: Nachweis der Superoxiddismutase-Aktivität (SOD) in BMDMs ( n = 5). C: Die Expression von Keap1 und Nrf2 wurde mittels Western Blot in BMDM-Lysaten analysiert und die Signalintensität quantifiziert ( n = 3). Die Daten sind als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * p < 0,05 vs. Kontrolle, # p < 0,05 vs. Hypoxie.
DiskussionUnsere Studie zeigt, dass Hypoxie die NLRX1-Expression im Lungengewebe und in Makrophagen von Mäusen herabreguliert. Wir liefern erstmals den Beweis, dass ein NLRX1-Mangel bei HPH-Mäusen zu einem signifikanten Umbau der Pulmonalarterien und einer Hypertrophie des rechten Ventrikels führt, begleitet von einem deutlichen Anstieg der Lungenmakrophagen und entzündungsfördernder Zytokine. Diese Ergebnisse legen nahe, dass die Schutzfunktion von NLRX1 bei HPH die Suppression der Makrophagenaktivität und -infiltration, möglicherweise durch negative Regulation von oxidativem Stress und NF-κB-Aktivierung, umfassen könnte.Pathologisch ist die HPH durch einen erhöhten pulmonalarteriellen Druck gekennzeichnet, der zu einer Hypertrophie des rechten Ventrikels führt. Die vaskuläre Remodellierung der Pulmonalarterie ist ein wichtiger auslösender Faktor in diesem Prozess [
27
]. Hypoxie induziert eine abnorme Proliferation und Migration von PASMCs, was zu einer fortschreitenden Lumeneinengung, einer verminderten Gefäßcompliance, einem erhöhten Gefäßwiderstand und der Entwicklung einer pulmonalen Hypertonie führt [
28
]. Gleichzeitig passt sich der rechte Ventrikel dem erhöhten pulmonalen Druck durch eine Steigerung der myokardialen Kontraktilität an, was zu einer Kardiomyozytenhypertrophie führt [
29
]. NLRX1, ein Mitglied der NLR-Familie, fungiert als Mustererkennungsrezeptor. Jüngste Erkenntnisse deuten darauf hin, dass NLRX1 eine negative regulatorische Rolle bei verschiedenen Infektions- und Autoimmunerkrankungen spielt. Eine Dysregulation der NLRX1-Expression oder -Aktivität verschlimmert häufig die Krankheitsprogression [
30
,
31
].In dieser Studie simulierten wir mithilfe einer Niederdruck-Sauerstoffkammer die hypoxischen Bedingungen in 5500 m Höhe und induzierten so eine pulmonale Hypertonie bei Mäusen. Nach der hypoxieinduzierten pulmonalen Hypertonie wurde in den Lungengeweben von Wildtyp-Mäusen (WT) eine verminderte NLRX1-Expression beobachtet. Histopathologische Untersuchungen zeigten, dass der Mangel an NLRX1 zu ausgeprägten pathologischen Veränderungen in Lunge und rechtem Ventrikel der Mäuse mit hypoxämischer pulmonaler Hypertonie (HPH) führte. Ein Vergleich zwischen WT- und NLRX1 <sup>−/−</sup> -Mäusen nach HPH-Induktion ergab ein erhöhtes Verhältnis von WT- zu WA-Prozentwerten in NLRX1<sup> −/−</sup> -Mäusen, was auf eine abnorme Verengung des pulmonalen Gefäßlumens hindeutet. Zusätzlich deutete ein deutlicher Anstieg der α-SMA-positiven Zellen auf eine abnorme Proliferation glatter Muskelzellen hin. Darüber hinaus wiesen NLRX1 -Knockout- Mäuse im Vergleich zu Wildtyp-Mäusen einen höheren rechtsventrikulären systolischen Druck (RVSP) und ein höheres Verhältnis von rechtsventrikulärem zu linksventrikulärem und systolischem Druck (RV/(LV + S)) auf. Zusammengenommen deuten unsere Ergebnisse auf eine mögliche Schutzfunktion von NLRX1 bei HPH hin. Pathologisch betrachtet verschlimmert das Fehlen von NLRX1 das Remodeling der Pulmonalarterie und die rechtsventrikuläre Hypertrophie, was zu einem erhöhten pulmonalarteriellen Druck führt.Längerer Sauerstoffmangel induziert oxidativen Stress und Entzündungen, Schlüsselfaktoren in der Entwicklung der pulmonalen Hypertonie, indem er die Proliferation und Migration von PASMCs und das nachfolgende Gefäßremodeling verstärkt. Sauerstoffmangel stört die Funktion der mitochondrialen Atmungskette und führt zur Bildung reaktiver Sauerstoffspezies (ROS) [
32
]. ROS können IKKα direkt oxidieren oder über TLR4 den NF-κB-Signalweg aktivieren und so eine anhaltende Entzündungsreaktion auslösen [
33
,
34
]. Die Entzündung wird durch NLRs moduliert, welche Multiproteinkomplexe bilden und Signalwege aktivieren, die in der Sekretion proinflammatorischer Zytokine wie IL-1β, IL-6 und IL-18 münden [
35
]. Studien legen nahe, dass NLRX1 zelluläre Prozesse indirekt moduliert, indem es den ROS-Spiegel kontrolliert und die TLR4-vermittelte NF-κB-Aktivierung durch Interaktion mit TRAF6 in aktivierten Immunzellen hemmt [
36
]. Dennoch bleibt der genaue Regulationsmechanismus von NLRX1 auf NF-κB unter hypoxischen Bedingungen unklar.Wir untersuchten den oxidativen Stress in der Lunge von Wildtyp- (WT) und NLRX1 - Knockout-Mäusen (NLRX1 −/−) durch Analyse der NF-κB-Aktivierung und der Konzentrationen entzündungsfördernder Zytokine. Die Analyse zeigte, dass NLRX1 −/−- Mäuse nach HPH im Vergleich zu WT-Mäusen signifikant erhöhte TAC- und SOD-Aktivitäten im Lungengewebe aufwiesen, begleitet von einer gesteigerten Nrf2-Aktivierung. Dies deutet auf eine erhöhte Produktion von Substanzen des oxidativen Stresses, einschließlich reaktiver Sauerstoffspezies (ROS), hin. Darüber hinaus waren die NF-κB-Signalaktivierung sowie die Produktion der Zytokine IL-1β und IL-6 im Lungengewebe von NLRX1 −/−- Mäusen deutlich höher als in dem von WT-Mäusen. Diese Ergebnisse legen eine zentrale Rolle von übermäßigem oxidativem Stress und Entzündungsreaktionen in der Pathogenese der schweren HPH mit NLRX1-Deletion nahe. Obwohl Nrf2- und NF-κB-Signalwege im Lungengewebe von NLRX1<sup> −/−</sup> -Mäusen während der hypoxisch-pulmonalen Hypertonie (HPH) gleichzeitig aktiviert waren, zeigte sich keine offensichtliche Korrelation zwischen ihnen. Wir berichteten bereits über einen signifikanten Unterschied in der Anzahl der Makrophagen im Lungengewebe von HPH-Mäusen zwischen Wildtyp- und NLRX1<sup> −/−</sup> -Gruppen. Es ist jedoch weiterhin unklar, ob NLRX1 die Hyperaktivierung von Makrophagen über einen ähnlichen Mechanismus fördert. Um diese Frage zu klären, führten wir eine Ko-Färbung von NLRX1 und F4/80 im Lungengewebe von Mäusen durch und stellten fest, dass Hypoxie die Expression von NLRX1 in pulmonalen Makrophagen herunterreguliert (Abbildung
S1
). Anschließend verwendeten wir primäre Knochenmark-abgeleitete Makrophagen (BMDMs) von NLRX1<sup> −/−</sup> -Mäusen und unsere In-vitro-Untersuchungen zeigten, dass NLRX1 in hypoxieinduzierten BMDMs herunterreguliert war. Zusammenfassend deuten unsere Ergebnisse darauf hin, dass Hypoxie oxidativen Stress auslöst und NF-κB/Nrf2 in Lungengewebe und Makrophagen aktiviert. Diese Reaktion wird durch einen NLRX1-Mangel deutlich verstärkt. Diese Verstärkung korreliert mit einer erhöhten Produktion von Entzündungszytokinen in vivo und in vitro. Die Ergebnisse legen nahe, dass die Aktivierung oder Hochregulierung von NLRX1 eine übermäßige Immunaktivierung in Makrophagen durch Modulation des oxidativen Stresses abschwächen könnte. Um die Schutzwirkung von NLRX1 unter hypoxischen Bedingungen weiter zu untersuchen, behandelten wir BMDMs mit NX-13, einem NLRX1-Agonisten, vor. Die Vorbehandlung mit NX-13 unterdrückte nicht nur den hypoxieinduzierten oxidativen Stress und die NF-κB-Aktivierung in BMDMs, sondern verringerte auch die Produktion von Entzündungszytokinen und die Nrf2-Aktivierung.NLRX1 moduliert bemerkenswerterweise auch andere wichtige Signalwege der pulmonalen Hypertonie (PH). Ein Mangel an NLRX1 verstärkt die NF-κB-Aktivierung und erhöht die Konzentration proinflammatorischer Zytokine in HPH-Mäusen, was mit seiner Rolle bei der Hemmung der NF-κB- und NLRP3-Inflammasom-Aktivierung übereinstimmt. Präklinische Studien haben gezeigt, dass NLRX1-KO-Mäuse unter Stress eine verstärkte kardiale Entzündung und NLRP3-Aktivierung aufweisen [
37
], was mit der Beobachtung übereinstimmt, dass ein NLRX1-Mangel die pulmonale Entzündung bei HPH verschlimmert. NLRX1 reguliert zudem die ROS-Produktion und die FUNDC1-vermittelte Mitophagie [
38
]. HPH ist durch übermäßige mitochondriale Fusion, gestörte Mitophagie und oxidativen Stress gekennzeichnet. Unsere Studie zeigt, dass ein Mangel an NLRX1 die ROS-Produktion, die Gesamt-Antioxidationskapazität (TAC), die SOD-Aktivität und die Nrf2-Aktivierung erhöht, während sein Verlust mitochondriale Defekte in Kardiomyozyten verschlimmert. Dies legt nahe, dass NLRX1 die pulmonale Hypertonie (HPH) durch mitochondriale Regulation verhindern kann. Indirekt zeigt die druckinduzierte Herzhypertrophie eine Herunterregulierung von NLRX1, und seine Überexpression stellt die mitochondriale Funktion wieder her, was den Befunden bei HPH entspricht [
39
]. Darüber hinaus zeigt der Human Protein Atlas, dass NLRX1 in humanen Lungenmakrophagen vorkommt, was mit unseren Beobachtungen an Mäusen übereinstimmt und die Relevanz für die menschliche Erkrankung unterstreicht [
40
]. Zusammenfassend zeigt unsere Studie, dass Hypoxie die NLRX1-Expression reduziert, was zu einer verstärkten Makrophageninfiltration, erhöhtem oxidativem Stress und einer Hyperaktivierung von NF-κB führt. Frühere Untersuchungen deuten darauf hin, dass NLRX1 die ROS-Produktion bei akuten Infektionen stimulieren und dadurch die angeborene Immunantwort verstärken kann [
41
,
42
,
43
]. Längerer Sauerstoffmangel führt jedoch zu anhaltendem oxidativem Stress und erhöhten Konzentrationen proinflammatorischer Mediatoren, was letztendlich zu chronischer Entzündung und pulmonaler Hypertonie führt (Abb.
10
). Daher fungiert NLRX1 wahrscheinlich als negativer Regulator der ROS-Bildung bei chronischer Entzündung. Um die Rolle von NLRX1 bei HPH zu klären, sind weitere Untersuchungen zur Aufklärung der zugrundeliegenden Mechanismen erforderlich.Abb. 10
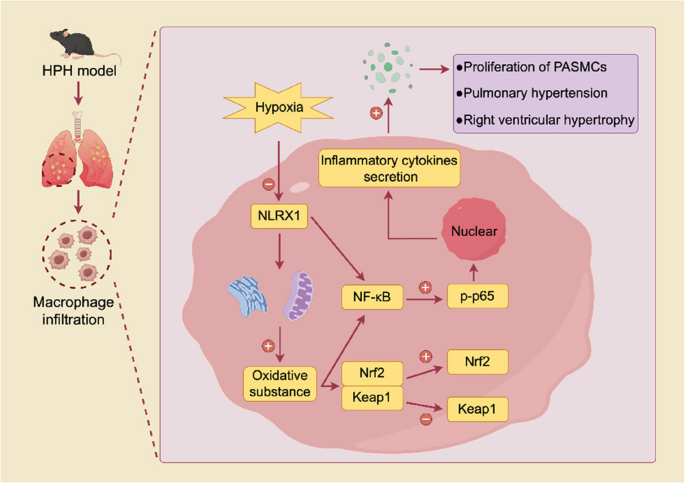 Der vorgeschlagene NLRX1-vermittelte Signalweg trägt zu oxidativem Stress und der Entzündungsreaktion bei HPH bei. Hypoxie reduziert die NLRX1-Expression in Makrophagen, was zu einer signifikanten Akkumulation im Lungengewebe von HPH-Mäusen führt. Die Herunterregulierung von NLRX1 verstärkt den oxidativen Stress in Makrophagen und fördert indirekt oder direkt die p65-Phosphorylierung. Darüber hinaus führt die verminderte Expression von Keap1 zur Aktivierung von Nrf2. Die anschließende p65-Phosphorylierung löst die Aktivierung des NF-κB-Signalwegs aus und fördert die Sekretion von Entzündungszytokinen. Diese Kaskade führt letztendlich durch die Stimulation der Proliferation von PASMCs zu pulmonaler Hypertonie und Rechtsherzhypertrophie (nach Figdraw).
AbschlussDie hypoxämische pulmonale Hypertonie (HPH) ist weiterhin eine lebensbedrohliche Erkrankung, die durch pulmonales Gefäßremodeling infolge hypoxiebedingter Entzündungen gekennzeichnet ist. Unsere Forschung zeigt, dass Hypoxie einen deutlichen Anstieg von Makrophagen im Lungengewebe auslöst, der mit einer signifikanten Reduktion des NLRX1-Spiegels einhergeht. Darüber hinaus verschlimmert das Fehlen von NLRX1 das pulmonale Gefäßremodeling, die pulmonale Hypertonie und die Rechtsherzhypertrophie bei Mäusen. In vitro führt der Mangel an NLRX1 unter hypoxischen Bedingungen zu verstärktem oxidativem Stress und einer Überaktivierung des NF-κB-Signalwegs in Makrophagen, was eine erhöhte Freisetzung von Entzündungszytokinen zur Folge hat. Die Vorbehandlung mit NX-13 mildert jedoch effektiv den oxidativen Stress, hemmt die NF-κB-Aktivierung und reduziert die nachfolgende Freisetzung von Entzündungszytokinen. Diese Ergebnisse legen nahe, dass NLRX1 eine negative regulatorische Rolle bei hypoxiebedingtem oxidativem Stress und der Entzündungsreaktion in Makrophagen spielt und somit ein potenzielles therapeutisches Ziel für die HPH darstellt.
Der vorgeschlagene NLRX1-vermittelte Signalweg trägt zu oxidativem Stress und der Entzündungsreaktion bei HPH bei. Hypoxie reduziert die NLRX1-Expression in Makrophagen, was zu einer signifikanten Akkumulation im Lungengewebe von HPH-Mäusen führt. Die Herunterregulierung von NLRX1 verstärkt den oxidativen Stress in Makrophagen und fördert indirekt oder direkt die p65-Phosphorylierung. Darüber hinaus führt die verminderte Expression von Keap1 zur Aktivierung von Nrf2. Die anschließende p65-Phosphorylierung löst die Aktivierung des NF-κB-Signalwegs aus und fördert die Sekretion von Entzündungszytokinen. Diese Kaskade führt letztendlich durch die Stimulation der Proliferation von PASMCs zu pulmonaler Hypertonie und Rechtsherzhypertrophie (nach Figdraw).
AbschlussDie hypoxämische pulmonale Hypertonie (HPH) ist weiterhin eine lebensbedrohliche Erkrankung, die durch pulmonales Gefäßremodeling infolge hypoxiebedingter Entzündungen gekennzeichnet ist. Unsere Forschung zeigt, dass Hypoxie einen deutlichen Anstieg von Makrophagen im Lungengewebe auslöst, der mit einer signifikanten Reduktion des NLRX1-Spiegels einhergeht. Darüber hinaus verschlimmert das Fehlen von NLRX1 das pulmonale Gefäßremodeling, die pulmonale Hypertonie und die Rechtsherzhypertrophie bei Mäusen. In vitro führt der Mangel an NLRX1 unter hypoxischen Bedingungen zu verstärktem oxidativem Stress und einer Überaktivierung des NF-κB-Signalwegs in Makrophagen, was eine erhöhte Freisetzung von Entzündungszytokinen zur Folge hat. Die Vorbehandlung mit NX-13 mildert jedoch effektiv den oxidativen Stress, hemmt die NF-κB-Aktivierung und reduziert die nachfolgende Freisetzung von Entzündungszytokinen. Diese Ergebnisse legen nahe, dass NLRX1 eine negative regulatorische Rolle bei hypoxiebedingtem oxidativem Stress und der Entzündungsreaktion in Makrophagen spielt und somit ein potenzielles therapeutisches Ziel für die HPH darstellt.
zu einer Hochregulation von Entzündungsmediatoren und fördert gleichzeitig oxidativen Stress sowie die Aktivierung des nukleären Faktors E2-verwandten Faktors 2 (Nrf2). Der Aktivator NX-13 von NLRX1 kann die Produktion von entzündungsfördernden Zytokinen in durch Hypoxie stimulierten BMDMs reduzieren und oxidativen Stress mindern.SchlussfolgerungenNLRX1 kann durch seine entzündungshemmenden und antioxidativen Eigenschaften hypoxiebedingte Lungenentzündungen unterdrücken und hypoxiebedingte pulmonale Hypertonie lindern. Daher könnte die gezielte Hochregulierung von NLRX1 eine neue Behandlungsstrategie für HPH darstellen.HintergrundDie hypoxische pulmonale Hypertonie (HPH) ist eine Erkrankung, die durch einen erhöhten pulmonalen Arteriendruck aufgrund einer längeren Exposition gegenüber einer hypoxischen Umgebung gekennzeichnet ist. Sie tritt häufig bei Personen auf, die in großen Höhen leben, und bei Patienten mit chronisch-obstruktiver Lungenerkrankung (COPD) [
1
]. Die pulmonale Gefäßremodellierung gilt allgemein als Hauptursache für den erhöhten pulmonalen Arteriendruck, während Hypoxie als zentraler Auslöser der HPH angesehen wird, da sie eine Entzündungsreaktion im Immunsystem stimuliert. Anhaltende Hypoxie induziert die Proliferation, Migration und phänotypische Veränderung von glatten Muskelzellen der Pulmonalarterie (PASMCs), was zu einer Verdickung der Gefäßwand und einer Verengung des Lumens führt [
2
]. Neuere Erkenntnisse deuten darauf hin, dass hypoxieinduzierte Entzündungen und metabolische Dysregulationen in Gefäßzellen zur Entwicklung der HPH beitragen. Perivaskuläre Entzündungen sind eng mit der Verdickung der Intima und Media der Gefäßwand verbunden [
3
]. Histopathologische Untersuchungen zeigen bei Patienten mit HPH entzündliche Infiltrate aus Makrophagen, Mastzellen und Neutrophilen, die pulmonale Gefäßläsionen umgeben [
4
]. Immunzellen interagieren nicht nur direkt mit pulmonalen Gefäßzellen, sondern setzen auch Entzündungsmediatoren frei, die die Proliferation von PASMCs fördern oder eine Endothelzellfunktionsstörung induzieren. Diese Prozesse treiben gemeinsam das Gefäßremodeling voran und führen zu einem erhöhten pulmonalen Arteriendruck.Die Familie der Nod-ähnlichen Rezeptoren (NLR) ist eine wichtige Gruppe von Mustererkennungsrezeptoren im menschlichen Körper, die primär die angeborene Immunantwort steuern und die innere Homöostase aufrechterhalten [
5
]. Unter diesen Rezeptoren nimmt NLRX1 eine herausragende Stellung ein. Er ist vorwiegend im Zytoplasma und in den Mitochondrien lokalisiert und spielt dort eine zentrale Rolle bei der Regulation von Immunität, Antioxidation und Apoptose [
6
]. Studien haben gezeigt, dass NLRX1 die Lipopolysaccharid-induzierte NF-κB-Aktivierung nachgeschaltet von Toll-like-Rezeptor 4 (TLR4) negativ reguliert, indem es in Fibroblasten oder Makrophagen mit dem TNF-Rezeptor-assoziierten Faktor 6 (TRAF6) interagiert und einen Komplex bildet [
7
]. In der T-Zell-Immunantwort moduliert NLRX1 die T-Zell-Aktivierung und -Differenzierung durch Beeinflussung der NF-κB-Signalübertragung [
8
,
9
]. NLRX1 besitzt bemerkenswerterweise eine Doppelfunktion in der Entzündungsreaktion und Immunregulation [
10
]. Es fördert Autophagie und Apoptose, um pathogene Mikroorganismen und Tumorzellen zu bekämpfen, und hemmt gleichzeitig Entzündungsprozesse, um Entzündungen zu lindern. Die zentrale Rolle von NLRX1 zeigt sich in der Pathogenese und Progression verschiedener Erkrankungen, darunter Virusinfektionen, Tumore und Autoimmunerkrankungen [
11
,
12
,
13
,
14
,
15
]. Obwohl die genaue Beteiligung von NLRX1 an der hypoxiebedingten pulmonalen Hypertonie (HPH) noch nicht vollständig erforscht ist, deuten bestehende Studien darauf hin, dass eine NLRX1-Supplementierung die Lungenentzündung deutlich reduzieren kann, wobei eine inverse Korrelation mit der Expressionsstärke entzündungsfördernder Zytokine besteht [
16
,
17
]. Diese Beobachtungen legen eine mögliche Rolle von NLRX1 bei der Linderung der hypoxiebedingten pulmonalen Hypertonie durch die Unterdrückung der Entzündungsreaktion im Lungengewebe nahe.Zusammenfassend lässt sich sagen, dass NLRX1 als Suppressor von NF-κB fungiert, einem zentralen Signalweg bei hypoxieinduzierter Entzündung und oxidativem Stress. Es wurde berichtet, dass NLRX1 die NF-κB-Signalübertragung in verschiedenen Zelltypen hemmt, was darauf hindeutet, dass seine entzündungshemmenden und antioxidativen Eigenschaften durch hypoxische Bedingungen beeinflusst werden könnten. Daher sind weitere Untersuchungen erforderlich, um den genauen Beitrag von NLRX1 im Kontext der Hypoxie-induzierten Hypoxie (HPH) aufzuklären.MethodenVersuchstiereWildtyp-Mäuse (WT) des Stammes C57BL/6 wurden vom Tierzentrum der Vierten Militärmedizinischen Universität (China) bezogen. NLRX1-Knockout-Mäuse (NLRX1 <sup>−/−</sup> ) des Stammes C57BL/6 wurden in Zusammenarbeit mit dem Institut für Mikrobiologie der Vierten Militärmedizinischen Universität generiert [
18
]. Beide Mäusestämme waren männlich und 6–8 Wochen alt. Die Protokolle dieser Studie wurden von der Ethikkommission für Tierversuche der Vierten Militärmedizinischen Universität genehmigt.Modellierung und Gruppierung von PH-MäusenWT- und NLRX1<sup> −/−</sup> -Mäuse wurden randomisiert in vier Gruppen eingeteilt: Kontrollgruppe, NLRX1<sup> −/− </sup>-Gruppe , Hypoxiegruppe und Hypoxie + NLRX1<sup> −/−</sup> -Gruppe (jeweils 20 Mäuse). Die Mäuse wurden für sechs Wochen in einer Unterdruckkammer (Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> , 10 % O <sub>2 </sub> ) oder unter Raumluftbedingungen (Kontrollgruppe, NLRX1 <sup>−/−</sup> , 21 % O <sub> 2</sub>) gehalten. Während der gesamten Studie hatten alle Mäuse sowohl unter Hypoxie- als auch unter Normoxiebedingungen uneingeschränkten Zugang zu Futter und Wasser.Hämodynamik und HypertrophiemessungDer systolische Druck im rechten Ventrikel (RVSP) von Mäusen wurde nach Pentobarbital-Natrium-Narkose gemessen. Die Rippen wurden chirurgisch freigelegt, und der Druckwandler wurde nach Anschluss an die intravenöse Nadel in den rechten Ventrikel kanüliert. Der Druck im rechten Ventrikel wurde mindestens 30 Sekunden lang kontinuierlich gemessen [
19
]. Alle Daten wurden mit dem Datenerfassungssystem Powerlab (AD Instruments) aufgezeichnet und analysiert. Im Anschluss an die Datenerfassung wurden die Mäuse durch sofortiges Ausbluten euthanasiert, und Lungen- und Herzgewebe wurden entnommen. Der rechte Ventrikel (RV) wurde vom linken Ventrikel (LV) einschließlich der Septumwand (S) getrennt, und beide wurden gewogen, um das Verhältnis des Gewichts der freien Wand des rechten Ventrikels zum Gewicht des linken Ventrikels plus Septum (RV/LV + S) zu berechnen [
20
].Histopathologische Untersuchung des rechten Ventrikels und der LungeDie aus den Mäusen entnommenen Lungen- und Herzgewebe wurden in Paraformaldehyd eingelegt und 24 Stunden lang fixiert. Anschließend wurden diese Gewebe in Wachsblöcke eingebettet, aus denen 5 μm dicke Schnitte angefertigt wurden. Die Gewebeschnitte wurden nach Hämatoxylin-Eosin-Färbung mikroskopisch untersucht. Die Gefäßwanddicke (WT), der äußere Gefäßdurchmesser (ED), die Gefäßwandfläche (WA) und die gesamte Gefäßfläche (TA) wurden gemessen, um WT% (WT/ED) und WA% (WA/TA) zu berechnen [21
]
. Die Dicke des rechten Ventrikels wurde mit einem Olympus-System gemessen.ImmunfluoreszenzDie Lungenschnitte wurden zunächst dreimal mit PBS gespült und anschließend durch Einlegen in Triton X-100 permeabilisiert. Nach Blockierung mit einer Immunfluoreszenz-Blockierungslösung (P0102, Beyotime) wurden die Schnitte über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-CD4 (ab133616, 1:500, Abcam), anti-F4/80 (29414-1-AP, 1:200, Proteintech), anti-α-SMA (A17910, 1:200, Abclonal) und anti-Nrf2 (A11159, 1:100, Abclonal). Anschließend erfolgte eine einstündige Inkubation mit sekundären Fluoreszenz-Antikörpern (ab150077, 1:500, ab150115, 1:200, Abcam) und eine 30-minütige Inkubation mit DAPI (C1005, Beyotime). Die Fluoreszenz wurde mittels Fluoreszenzmikroskopie abgebildet und die Daten mit Caseviewer verarbeitet.Isolierung und Kultivierung von aus dem Knochenmark stammenden Makrophagen (BMDMs)Für die Isolierung von Knochenmarkzellen wurden WT- und NLRX1 −/−- Mäuse im Alter von 4 bis 6 Wochen ausgewählt [
22
]. Nach der Erythrozytenlyse wurden die extrahierten Knochenmarkzellen in 2 ml PBS resuspendiert. Anschließend wurden die Zellen in RPMI 1640 mit 10 % FBS (10099-141, Gibco) und einer Konzentration von 10 ng/ml m-CSF (HZ-1192, Proteintech) überführt. Nach 24-stündiger Inkubation wurde die Suspension in 6-Well-Platten pipettiert. Die Proliferation und Differenzierung der BMDMs erstreckte sich über eine Woche, sodass das Medium an den Tagen 2, 4 und 6 erneuert werden musste.Zellbehandlung und Gruppierung von BMDMsWT- und NLRX1<sup> −/−</sup> -BMDMs wurden randomisiert in fünf Gruppen eingeteilt: Kontrolle, NLRX1 <sup>−/−</sup> , Hypoxie, Hypoxie + NLRX1<sup> −/−</sup> und Hypoxie + NX-13. NX-13 (HY-141521, MedChemExpress) wurde in DMSO gelöst und auf eine Endkonzentration von 0,5 µM eingestellt [
8
,
23
]. Die Hypoxie-Gruppen wurden 24–48 h lang hypoxisch in einem Inkubator mit 5 % O <sub>2</sub> , die Normoxie-Gruppen in einem Inkubator mit 21 % O <sub>2 </sub> kultiviert . Anschließend wurden Zellproben für weitere Experimente entnommen.RNA-Extraktion und -NachweisRNA wurde aus Lungengewebe und BMDMs mit dem RNA-Extraktionskit (R0026, Beyotime) isoliert. Die RNA-Konzentration wurde gemessen und verdünnt. Anschließend erfolgte die cDNA-Synthese mit dem SweScript RT cDNA-Synthesekit (G3333, Servicebio). Das TB Green PCR-Kit (G3320, Servicebio) wurde den Proben zugegeben. Die quantitative Echtzeit-PCR (qRT-PCR) wurde auf einem PCR-Gerät von Biosystems durchgeführt. Die Genexpression wurde im Vergleich zu den Kontrollen mit der 2<sup>-ΔΔCt</sup>-Formel analysiert. Die Expression wurde auf die mittlere Expression aller Kontrollprobanden normiert.Folgende Primer wurden verwendet: β-Actin, 5'-CACGATGGAGGGGCCGGACTCATC-3' (vorwärts) und 5' -TAAAGACCTCTATGCCAACACAGT-3' (rückwärts); NLRX1, 5'-CAGATTGGTAACAAAGGAGCCA-3' (vorwärts) und 5'-CGTTCGGTTTATCTTCAGAGCA-3' (rückwärts).Western BlotGesamtproteine wurden aus Lungengewebe und BMDMs isoliert und die Konzentration mittels BCA-Assay-Kit (P0012, Beyotime) quantifiziert. Proben mit 50 µg Protein wurden auf ein 10%iges SDS-PAGE-Gel aufgetragen und einer einstündigen Gelelektrophorese unterzogen. Anschließend wurden die Proteine auf PVDF-Membranen transferiert und mit Western-Blot-Blockierungslösung (P30500, NCM Biotech) behandelt. Die Membranen wurden über Nacht bei 4 °C mit folgenden Antikörpern inkubiert: anti-NLRX1 (17215-1-AP, 1:1000, Proteintech), anti-IL-1β (A16288, 1:1000, Abclonal), anti-IL-18 (A1115, 1:1000, Abclonal), anti-pNF-κB p65 (82335-1-RR, 1:1000, Proteintech), anti-NF-κB p65 (10745-1-AP, 1:1000, Proteintech), anti-Nrf2 (A0674, 1:1000, Abclonal), anti-Keap1 (10503-2-AP, 1:1000, Proteintech) und anti-β-Actin (ab8227, 1:5000, Abcam). Inkubation mit dem Sekundärantikörper (RGAR001, 1:5000, Proteintech) für 1 h. Der Nachweis erfolgte mittels Chemilumineszenzsystem (CLINX ChemiScope).ZytokinmessungNach Aspiration und Zentrifugation des Überstands aus der BMDM-Kultur wurde die Konzentration von IL-1β und IL-6 mittels ELISA-Kits (R&D) bestimmt. Die Gebrauchsanweisung wurde vom Hersteller bereitgestellt.Messung der Superoxiddismutase (SOD)-AktivitätDie Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die Gesamt-SOD-Aktivität im Überstand mithilfe des Gesamt-SOD-Assay-Kits (S0101S, Beyotime) gemäß den Herstellerangaben bestimmt.Messung der gesamten antioxidativen Kapazität (TAC)Die Proteinkonzentrationen in den Überständen von Lungengewebe und BMDM-Homogenaten wurden nach Zentrifugation gemessen. Anschließend wurde die antioxidative Gesamtkapazität (TAC) im Überstand mithilfe des Testkits zur Bestimmung der antioxidativen Gesamtkapazität (S0121, Beyotime) gemäß den Herstellerangaben bestimmt.Statistische AnalysenDie Ergebnisse wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt und mit GraphPad Prism 9.0 analysiert. Signifikante Unterschiede zwischen den beiden Gruppen wurden mittels t-Test nach Student ermittelt. Unterschiede zwischen mehreren Gruppen wurden durch eine einfaktorielle Varianzanalyse (ANOVA) mit gegebenenfalls anschließendem Bonferroni-Post-hoc-Test untersucht. Ein p-Wert < 0,05 wurde als statistisch signifikant angesehen.ErgebnisseHypoxie führte zu einer verminderten Expression von NLRX1 im Lungengewebe von Mäusen.NLRX1 ist für seine negative Regulation von Entzündungen und Oxidation bekannt. Der Einfluss von Hypoxie auf die NLRX1-Funktion wurde jedoch bisher nicht untersucht. Daher analysierten wir die NLRX1-Expression im Lungengewebe von Mäusen mit HPH. Unsere Ergebnisse (Abb.
1
A und zeigen eine Reduktion sowohl der mRNA- als auch der Proteinspiegel von NLRX1 im Lungengewebe von HPH-Mäusen im Vergleich zur Kontrollgruppe.Abb. 1
. Anschließend führten wir eine Immunfluoreszenzfärbung durch, um α-Aktin glatter Muskulatur (α-SMA) in Lungenschnitten zu markieren. Die Ergebnisse zeigten eine erhöhte Anzahl α-SMA-positiver Zellen, die die Lungengefäße in NLRX1<sup> −/−</sup> -Mäusen mit HPH im Vergleich zu Wildtyp-Mäusen umgeben, was die Verschlimmerung des Gefäßumbaus weiter belegt (Abb.
2C
).Abb. 2
. C Immunfluoreszenzaufnahmen von Lungenschnitten, gefärbt für α-SMA (rot) und DAPI (blau), mit Quantifizierung ( n = 5). Maßstabsbalken: 50 μm. D Repräsentative Bilder der RVSP-Kurve mit Quantifizierung ( n = . E Herzgewebe wurde nach Hämatoxylin-Eosin-Färbung histologisch mittels Mikroskop analysiert. Maßstabsbalken: 250 μm. F RV/(LV + S) wurde zur Analyse der rechtsventrikulären Hypertrophie berechnet ( n = . Die Daten wurden als Mittelwert ± Standardfehler des Mittelwerts (SEM) dargestellt. * P < 0,05 vs. Kontrolle, ● P < 0,05 vs. NLRX1 −/− , # P < 0,05 vs. Hypoxie
NLRX1-vermittelte hämodynamische Reaktion und rechtsventrikuläre Hypertrophie bei HPH-MäusenHämodynamische Veränderungen, die primär auf vaskuläres Remodeling zurückzuführen sind, sind ein wichtiger Indikator für pulmonale Hypertonie. Die Gefäßdynamik wird durch die Messung des rechtsventrikulären systolischen Drucks (RVSP) beurteilt. Wie in Abb.
2D
dargestellt , führte die Exposition gegenüber Hypoxie zu einem signifikanten Anstieg des RVSP bei Mäusen mit pulmonaler Hypertonie (HPH). Insbesondere war der RVSP bei NLRX1<sup> −/−</sup> -Mäusen im Vergleich zu Wildtyp-Mäusen (WT) erhöht (Abb.
2D
). Diese hämodynamischen Veränderungen können den rechten Ventrikel zusätzlich belasten und letztendlich eine Rechtsherzhypertrophie verursachen. Die histologische Analyse zeigte eine Verdickung des rechten Ventrikels bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe, wobei die Hypertrophie bei NLRX1<sup> −/−</sup> -Mäusen stärker ausgeprägt war als bei WT-Mäusen (Abb.
2E
). Das Verhältnis RV/LV + S ist ein häufig verwendeter Indikator für die Rechtsherzhypertrophie. Die Ergebnisse zeigten einen Anstieg des RV/LV + S-Verhältnisses bei HPH-Mäusen im Vergleich zur Normoxie-Gruppe. Unter hypoxischen Bedingungen wurden bei NLRX1<sup> −/−</sup> -Mäusen sogar noch höhere Werte beobachtet als bei Wildtyp-Mäusen (Abb.
2F
). Zusammenfassend deuten diese Befunde darauf hin, dass die Deletion von NLRX1 die HPH bei Mäusen verschlimmert.Ein NLRX1-Mangel führte bei HPH-Mäusen zu einer Hochregulierung von Entzündungsmediatoren durch Aktivierung von NF-κB im Lungengewebe.Hypoxie stimuliert bei pulmonaler Hypertonie primär Entzündungsreaktionen durch die Induktion der Produktion von Entzündungszytokinen wie IL-1β und IL-18. Um die Rolle von NLRX1 bei hypoxieinduzierter Entzündung zu untersuchen, bestimmten wir die Konzentrationen dieser Entzündungszytokine im Lungengewebe von Mäusen jeder Versuchsgruppe. Die IL-1β- und IL-18-Konzentrationen waren im Lungengewebe von Mäusen mit pulmonaler Hypertonie im Vergleich zur Normoxie-Gruppe erhöht. Darüber hinaus waren die Konzentrationen dieser Zytokine in NLRX1<sup> −/− </sup>-Mäusen mit pulmonaler Hypertonie signifikant höher als in Wildtyp-Mäusen (Abb.
3A
).Abb. 3
.Abb. 4
.Abb. 6
. Die anschließende Analyse der Keap1- und Nrf2-Expression in BMDMs ergab, dass die Keap1-Expression in WT-BMDMs nach 24 h Hypoxie abnahm, während die Nrf2-Expression im Vergleich zur Normoxie-Gruppe anstieg, ohne dass nach 48 h signifikante Unterschiede festgestellt wurden. Wichtig ist, dass die Deletion von NLRX1 zu jedem Zeitpunkt im Vergleich zu WT-BMDMs und der Normoxie-Gruppe deutlichere Veränderungen der Keap1- und Nrf2-Expression in hypoxieinduzierten BMDMs hervorrief (Abb.
7
C).Abb. 7
. Darüber hinaus untersuchten wir die Aktivierung von NF-κB in BMDMs und beobachteten eine verminderte Phosphorylierung von p65 in hypoxieinduzierten BMDMs zu jedem Zeitpunkt nach NX-13-Vorbehandlung (Abb.
8
C).Abb. 8
. Frühere Untersuchungen unserer Gruppe zeigten, dass das Fehlen von NLRX1 die Nrf2-Aktivierung in Lungengewebe und BMDMs von HPH-Mäusen verstärkt. Anschließend bestimmten wir die Keap1- und Nrf2-Spiegel in BMDMs. Interessanterweise stellte die NX-13-Vorbehandlung die Keap1-Expression nach 24 h Hypoxieexposition wieder her, reduzierte jedoch die Nrf2-Expression zu jedem Messzeitpunkt (Abb.
9
C). Aus den Ergebnissen von Tier- und Zellstudien schließen wir, dass NX-13 oxidativen Stress und die NF-κB-Aktivierung durch die Stimulation von NLRX1 in Makrophagen abschwächen und dadurch die Zellaggregation verringern sowie die Produktion von entzündungsfördernden Zytokinen reduzieren kann.Abb. 9
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.