
- Beiträge: 1757
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Therapeutische Herausforderungen und neue therapeutische Ziele bei kombinierter
Therapeutische Herausforderungen und neue therapeutische Ziele bei kombinierter
19 Sep 2025 13:58 #2395
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Therapeutische Herausforderungen und neue therapeutische Ziele bei kombinierter wurde erstellt von danny
Therapeutische Herausforderungen und neue therapeutische Ziele bei kombinierter kapillärer pulmonaler Hypertonie: eine ÜbersichtDie kombinierte prä- und postkapilläre pulmonale Hypertonie (Cpc-PH) tritt häufig auf und hat eine schlechte Prognose. Sie ist ein bedeutender Subtyp der pulmonalen Hypertonie, der mit einer linksseitigen Herzerkrankung (PH-LHD) einhergeht. Die komplizierte Pathophysiologie der Cpc-PH ist vor allem durch einen erhöhten Lungenvenendruck gekennzeichnet, der zu einem Anstieg des retrokapillären Drucks führt, gefolgt von einem erhöhten Lungenarteriendruck und einem deutlichen Anstieg des pulmonalvaskulären Widerstands (PVR). Für die Cpc-PH gibt es derzeit keinen klar definierten Behandlungsplan und es sind zahlreiche Hindernisse zu überwinden. Bei Patienten mit Cpc-PH ist die Wirksamkeit gezielter Medikamente gegen pulmonale Hypertonie begrenzt und umstritten. Neuere Forschungen haben ergeben, dass die Prävalenz und das Fortschreiten der Cpc-PH durch genetische Faktoren, das metabolische Syndrom, oxidativen Stress und Fibrose beeinflusst werden können. Um Ärzten dabei zu helfen, Patienten mit Cpc-PH besser zu betreuen und zu behandeln, bietet dieser Bericht eine detaillierte Beschreibung der Epidemiologie, Pathogenese, Diagnosetechniken, des aktuellen Behandlungsstatus und der potenziellen Therapieziele der Krankheit. EinführungDie europäischen Leitlinien für die Diagnose und Behandlung der pulmonalen Hypertonie (PH) von 2022, die ESC/ERS-Leitlinien 2022 für die Diagnose und Behandlung der pulmonalarteriellen Hypertonie, definieren PH als den mittleren pulmonalarteriellen Druck (mPAP), gemessen durch Rechtsherzkatheterisierung (RHC) auf Meereshöhe und in Ruhe ≥20 mmHg, und den pulmonalarteriellen Verschlussdruck (PAWP) > 15 mmHg (
1
). Im Vergleich zu den Leitlinien von 2015 wurde der Schwellenwert für mPAP von 25 mmHg auf 20 mmHg gesenkt und der Schwellenwert für PVR von 3 WU auf 2 WU, was der Früherkennung und Diagnose von Patienten mit PH förderlicher ist. Pulmonalarterielle Hypertonie in Verbindung mit einer linksseitigen Herzerkrankung (PH-LHD) ist die häufigste Form und macht etwa 68,5 % aller PH-Fälle aus (
2
). PH-LHD wird in zwei Subtypen unterteilt: Isolierte postkapilläre PH (Ipc-PH): mPAP > 20 mmHg, PAWP > 15 mmHg, PVR ≤ 2 WU; Kombinierte prä- und postkapilläre PH (Cpc-PH): mPAP > 20 mmHg, PAWP > 15 mmHg, PVR > 2 WU (
3
). Die kombinierte prä- und postkapilläre pulmonale Hypertonie (Cpc-PH) ist ein spezifischer Phänotyp der mit einer linksseitigen Herzerkrankung verbundenen pulmonalen Hypertonie (PH-LHD, WHO-Kategorie 2), der durch das gleichzeitige Auftreten von retrokapillärer Hypertonie (erhöhter pulmonalvenöser Druck) und pulmonalvaskulärer Umgestaltung (präkapilläre Hypertonie) aufgrund einer linksseitigen Herzerkrankung gekennzeichnet ist.In der Hämodynamik wird Cpc-PH normalerweise mit einer Linksherz- oder Lungenerkrankung in Verbindung gebracht, die zu erhöhtem pulmonalvenösen Druck führt und in der Folge den pulmonalarteriellen Druck beeinflusst. Im Frühstadium ist der erhöhte PAP hauptsächlich auf die passive Übertragung des linken Vorhofdrucks zurückzuführen. In diesem Stadium bleibt der PVR normalerweise im Normbereich und hat noch keine signifikanten Veränderungen der Lungengefäßstruktur verursacht. Wenn der linksatriale Druck weiter ansteigt, beginnen Veränderungen in der Lungengefäßstruktur aufzutreten: Venen werden durchlässiger, es kommt zu Kollagenablagerungen, was zu einer Verdickung der Gefäßwände und einer Verengung des Lumens führt; Kapillaren und kleine Arterien entwickeln unter langfristiger Hochdruckstimulation eine Intimafibrose und eine mediale Hypertrophie der Lungenarterien, was letztendlich zu einem Anstieg des PVR führt. Wenn der PVR 2 WU übersteigt, wird Cpc-PH etabliert. Die Wahrscheinlichkeit eines Krankenhausaufenthalts wegen Herzinsuffizienz oder eines Todes jeglicher Ursache stieg mit jedem Anstieg des mPAP um 1 mmHg um 2 % und mit jedem Anstieg des PVR um 1 WU um 7 % (
4
). Der neuen Definition zufolge liegt der ideale PVR-Schwellenwert bei knapp 2 WU bzw. 2,2 WU. Die Diagnose und Behandlung der kombinierten kapillären pulmonalen Hypertonie ist schwierig. Es muss eine individuelle Behandlungsstrategie entwickelt werden, die die Auswirkungen der Lungen- und Linksherzerkrankung berücksichtigt (
Abbildung 1
). Dies ist von entscheidender Bedeutung für das Verständnis des pathophysiologischen Prozesses der pulmonalen Hypertonie und die Verbesserung der Patientenergebnisse. Abbildung 1
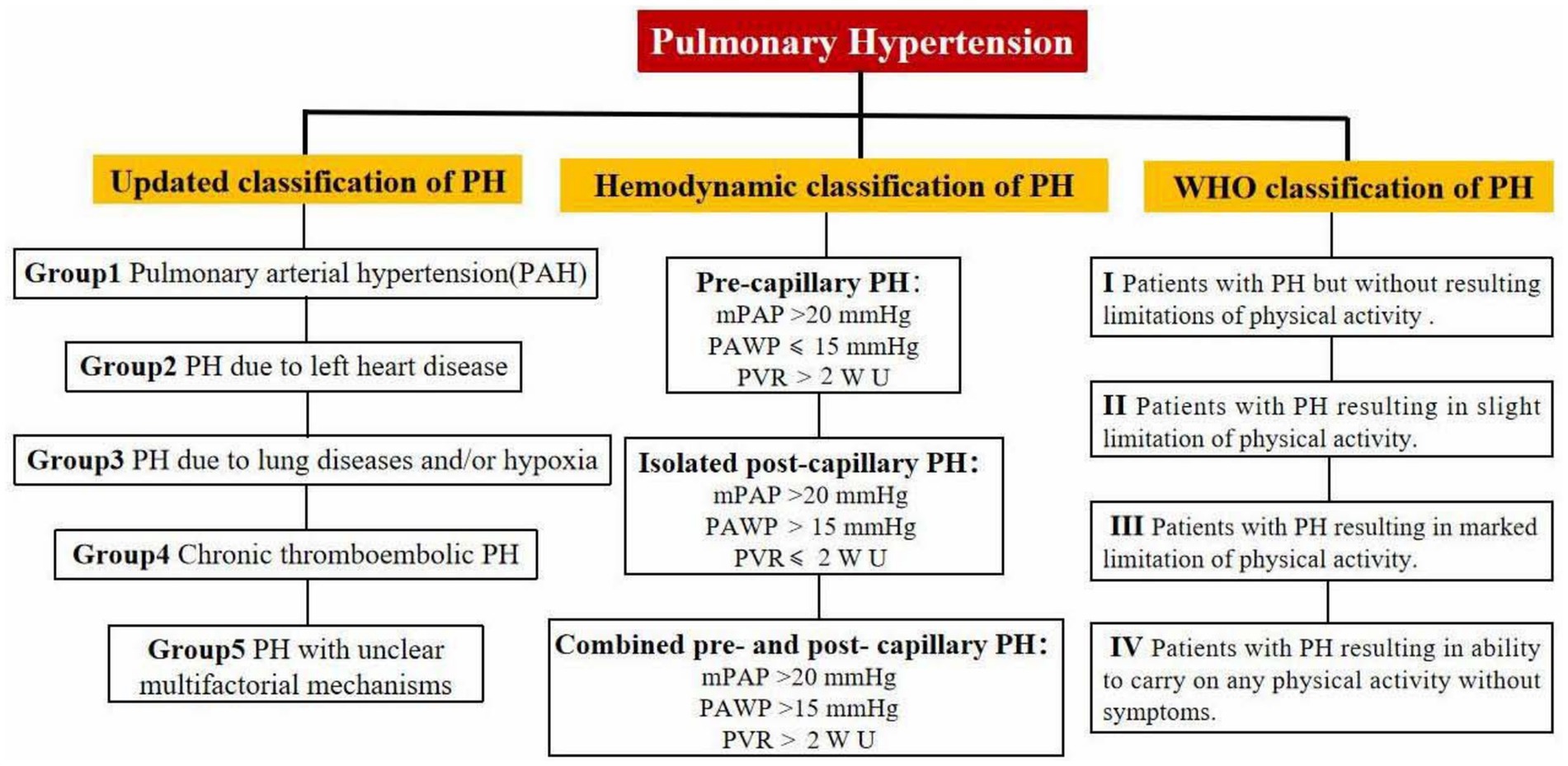 Abbildung 1. WHO-Klassifikation, ätiologische Klassifikation und hämodynamische Klassifikation der pulmonalen arteriellen Hypertonie. EpidemiologieWeltweit leidet etwa 1 % der Menschen an pulmonaler Hypertonie, und bei den über 65-Jährigen steigt die Häufigkeit auf 10 %. In Industrieländern (z. B. Europa und den USA) liegt die jährliche Inzidenz von Cpc-PH bei etwa 5–10 Fällen pro 100.000 Personen, was 5–10 % aller neuen Fälle von pulmonaler Hypertonie entspricht. Das Erkrankungsalter ist bei älteren Menschen (> 65 Jahre) höher (
1
–
5
), was mit einer hohen Inzidenz von Linksherzerkrankungen einhergeht, und der Anteil der Frauen ist etwas höher. Hypertonie (> 70 %), Diabetes und Fettleibigkeit (BMI > 30 %) sind die Hauptrisikofaktoren bei Patienten mit Grunderkrankungen, und die Inzidenz von Cpc-PH ist bei Patienten mit Herzklappenerkrankungen (z. B. Mitralklappeninsuffizienz) und Vorhofflimmern höher. Die Sterblichkeit ist bei Patienten mit Cpc-PH wesentlich höher als bei Patienten mit PH-LHD allein (die 2-Jahres-Überlebensrate beträgt etwa 50 – 60 %, die 5-Jahres-Überlebensrate etwa 40 – 50 % gegenüber 70 – 80 %) und das Sterberisiko steigt mit jeder Erhöhung des PVR um 1 WU um 20 – 30 % (
3
–
6
). In Studien zu verschiedenen Arten von pulmonaler Hypertonie war ein leicht erhöhter mPAP (21 – 24 mmHg) signifikant mit einer erhöhten Sterblichkeit verbunden (
3
–
7
). Mehrere Studien an Patienten mit systemischer Sklerose haben gezeigt, dass Patienten mit 24 mmHg ≥ mPAP ≥ 21 mmHg nicht nur eine höhere Sterblichkeit haben, sondern mit größerer Wahrscheinlichkeit eine pulmonale Hypertonie (mPAP ≥ 25 mmHg) entwickeln (
3
). Insgesamt entwickelten 5 – 12 % der Patienten mit linksseitiger Herzerkrankung (PH-LHD) eine Cpc-PH. EU-US- und US-Kohortenstudien zufolge entwickelten 1,5 bis 3 % der Patienten mit Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF) auch eine Cpc-PH; und zwischen 2 und 5 % der Patienten mit Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) entwickelten eine Cpc-PH (aufgrund einer Lungengefäßerkrankung und des allgemeinen metabolischen Syndroms). Pulmonale Hypertonie ist mit einer zweifach erhöhten Mortalität bei Menschen mit HFrEF und HFpEF verbunden. Die 3- und 5-Jahres-Sterblichkeitsraten für PH-LHD lagen bei 42,3 bzw. 52,6 % in einer Kohortenstudie mit 4.621 Patienten mit dieser Erkrankung (
8
). Alle diese klinischen Kohortenstudien zu PH-LHD stammen jedoch aus westlichen Ländern. Die jährliche Konversionsrate von Ipc-PH zu Cpc-PH beträgt etwa 3 bis 6 % (
9
). Bei bestimmten Personengruppen mit systemischer Sklerose können eine Linksherzerkrankung und eine PAH gleichzeitig vorliegen, was das Risiko einer Cpc-PH erhöht. Ältere Menschen, Diabetiker (30–40 %) und Patienten mit chronischer Nierenerkrankung (GFR < 60 ml/min/1,73 m 2 ) haben häufiger einen gemischten pH-Wert (
10
).In China fehlen derzeit repräsentative Daten zu PH-LHD, insbesondere in Form von Langzeit-Follow-up-Berichten. Daher sind multizentrische Registrierungsstudien und die Erstellung einer PH-LHD/Cpc-PH-Datenbank auf der Grundlage von RHC als Standarddiagnose in China dringend erforderlich.Pathophysiologische MechanismenCpc-PH beinhaltet komplexe pathophysiologische Mechanismen. Der Anstieg des PAWP spiegelt eine Funktionsstörung des linken Vorhofs wider. Dieser Druckanstieg überträgt sich auf den Lungenkreislauf und führt zu einem Anstieg von PAP und PVR sowie einer Abnahme der Pulmonalarterien-Compliance (PAC). Dies führt schließlich zu einer endothelialen Dysfunktion, einem pulmonalvaskulären Umbau und einer weiteren Erhöhung der Nachlast des rechten Ventrikels.Mechanismen der pulmonalen Gefäßumgestaltung und FibroseDas zentrale pathologische Merkmal der Cpc-PH ist der Umbau der Lungengefäße. Er ist gekennzeichnet durch Intimaproliferation, Mediahypertrophie und Adventitiafibrose der Lungenarteriolen. In den frühen Stadien führt ein erhöhter Füllungsdruck im linken Ventrikel zu einer Beeinträchtigung des pulmonalvenösen Rückflusses und einem erhöhten Lungengefäßdruck, jedoch zu keinen signifikanten strukturellen Veränderungen. Diese Erkrankung wird als IPC-PH (passive PH) bezeichnet (
11
). Im weiteren Krankheitsverlauf kommt es zu strukturellen und funktionellen Veränderungen der Lungenarterien mit Verdickung und Verhärtung der Gefäßwände, Einengung des Lumens, erhöhtem Widerstand und dauerhaft erhöhtem Lungengefäßwiderstand, was zu Rechtsherzinsuffizienz und dem Eintritt in die Cpc-PH-Phase führt (
12
). Bei Patienten mit Cpc-PH kommt es häufiger zu Funktionsstörungen des rechten Ventrikels und zu einem Umbau der Pulmonalarterie, mit signifikanten Merkmalen eines hypertrophen Umbaus der distalen Pulmonalarterie, einer Fibrose und eines Lumenverschlusses (
13
,
14
).Makrophagen interagieren mit pulmonalvaskulären Endothelzellen und treiben so den pulmonalvaskulären Umbau voran. Dabei kommt es zu einer Zunahme von CD68 + -Zellen (Makrophagen-Subtyp M2) in der Adventitiaschicht. Adventitiafibroblasten könnten die primären Mediatoren ihrer Aktivierung und Polarisierung sein. Das Fettsäure-bindende Protein 5 (FABP5) verschlimmert die pulmonalarterielle Fibrose durch Aktivierung des Wnt/ β -Catenin-Signalwegs, der in Cpc-PH-Mausmodellen signifikant hochreguliert ist und eng mit der pulmonalarteriellen Fibrose verwandt ist. In-vitro- Experimente haben gezeigt, dass die Hemmung von FABP5 (mittels siRNA oder Antagonisten) TGF-β1-induzierte fibrotische Reaktionen abschwächen kann. Spezifische alveoläre Kapillarendothelzellen (Plvap + ) können das mesenchymale Gen aSMA vorübergehend aktivieren und so die Umgestaltung der extrazellulären Matrix, die Gefäßintegrität und die Zell-Zell-Interaktionen beeinflussen (
15
,
16
).SBFI-26 ist ein selektiver FABP5-Inhibitor, der Fibrose durch Modulation des Wnt/ β -Catenin-Signalwegs hemmen kann. Dadurch werden Fibrose und Remodellierung der Lungenarterie in Tiermodellen signifikant unterdrückt und die Wanddicke der Lungenarterie sowie die Kollagenablagerung reduziert. Die zirkuläre RNA circALMS1 ist bei Patienten mit pulmonaler arterieller Hypertonie herunterreguliert, und ihre Überexpression kann die Proliferation und Migration pulmonaler mikrovaskulärer Endothelzellen durch Unterdrückung des miR-17-3p/YTHDF2-Signalwegs hemmen und so die Rechtsherzfunktion verbessern (
17
,
18
).Neurohumorale MechanismenDazu gehören sympathische Überaktivierung, Aktivierung des Renin-Angiotensin-Aldosteron-Systems (RAAS), entzündliche Zellinfiltration und Reaktionen auf oxidativen Stress. Angiotensin II (Ang II) aktiviert den RhoA/Rho-Kinase-Signalweg, hemmt die Aktivität der Myosin-Leichtkettenphosphatase (MLCP) und der endothelialen Stickoxidsynthase (eNOS), was zu Vasokonstriktion und endothelialer Dysfunktion führt (
19
). Die ACE2-Ang (
1
–
7
)-Mas-Achse fördert die Freisetzung von Stickoxid (NO) und Prostaglandinen und übt vasodilatatorische, antiproliferative und entzündungshemmende Effekte aus, wobei eine erhöhte Aktivität die pulmonale Hämodynamik verbessert (
20
). Rekombinantes humanes lösliches ACE2 (rhACE2) kann den pulmonalarteriellen Druck verbessern und oxidativen Stress reduzieren (
21
,
22
).Funktionsstörung des linken VorhofsHFpEF, HFrEF und VHD können im Rahmen des Umbauprozesses zu erhöhtem Druck im linken Vorhof (LAP), erhöhtem Volumen und anschließender Vergrößerung des linken Vorhofs, beeinträchtigter Kontraktilität und interstitieller Fibrose führen (
23
). Der Umbau des linken Vorhofs schwächt dessen Barrierefunktion, wodurch passiv Druck auf die Lungengefäße übertragen wird, was zu erhöhtem Lungenvenendruck und Lungenstauung führt (
24
). Plötzliche Erhöhungen des LAP können zu einem „alveolarkapillären Spannungsversagen“ führen, wodurch die alveolarkapilläre Barriere zerstört wird und ein Lungenödem verursacht wird (
25
). Unter dem Einfluss einer endothelialen Dysfunktion, neurohumoraler Faktoren und einer Infiltration entzündlicher Zellen führen anhaltende Veränderungen des LAP zu strukturellen Anomalien der Lungengefäße und einem erhöhten pulmonalvaskulären Widerstand (
26
–
28
).Pulmonale vaskuläre endotheliale DysfunktionEine Schädigung der Endothelzellen führt zu Vasokonstriktion, Entzündungsreaktionen und Fibrose. Vasoreaktive Substanzen wie Stickstoffmonoxid (NO), Prostacyclin (PGI2) und Endothelin-1 (ET-1) regulieren gemeinsam die pulmonale Vasodilatation und -konstriktion (
29
). NO wird von eNOS synthetisiert und freigesetzt, wodurch die Blutgefäße über den NO-sGC-cGMP-PKG-Signalweg erweitert, die Proliferation glatter Muskelzellen gehemmt und der pulmonale Gefäßumbau verlangsamt wird (
30
). Erhöhte Aldosteronspiegel können oxidativen Stress auslösen, die ET-B-Rezeptorsignalisierung beeinträchtigen, die NO-Synthese und Bioverfügbarkeit verringern. Gleichzeitig kann eine übermäßige ET-1-Produktion die eNOS-Expression hemmen und so die NO-Sekretion verringern (
31
,
32
).Entzündungs- und ChemotaxismechanismenCXCL8 (IL-
Abbildung 1. WHO-Klassifikation, ätiologische Klassifikation und hämodynamische Klassifikation der pulmonalen arteriellen Hypertonie. EpidemiologieWeltweit leidet etwa 1 % der Menschen an pulmonaler Hypertonie, und bei den über 65-Jährigen steigt die Häufigkeit auf 10 %. In Industrieländern (z. B. Europa und den USA) liegt die jährliche Inzidenz von Cpc-PH bei etwa 5–10 Fällen pro 100.000 Personen, was 5–10 % aller neuen Fälle von pulmonaler Hypertonie entspricht. Das Erkrankungsalter ist bei älteren Menschen (> 65 Jahre) höher (
1
–
5
), was mit einer hohen Inzidenz von Linksherzerkrankungen einhergeht, und der Anteil der Frauen ist etwas höher. Hypertonie (> 70 %), Diabetes und Fettleibigkeit (BMI > 30 %) sind die Hauptrisikofaktoren bei Patienten mit Grunderkrankungen, und die Inzidenz von Cpc-PH ist bei Patienten mit Herzklappenerkrankungen (z. B. Mitralklappeninsuffizienz) und Vorhofflimmern höher. Die Sterblichkeit ist bei Patienten mit Cpc-PH wesentlich höher als bei Patienten mit PH-LHD allein (die 2-Jahres-Überlebensrate beträgt etwa 50 – 60 %, die 5-Jahres-Überlebensrate etwa 40 – 50 % gegenüber 70 – 80 %) und das Sterberisiko steigt mit jeder Erhöhung des PVR um 1 WU um 20 – 30 % (
3
–
6
). In Studien zu verschiedenen Arten von pulmonaler Hypertonie war ein leicht erhöhter mPAP (21 – 24 mmHg) signifikant mit einer erhöhten Sterblichkeit verbunden (
3
–
7
). Mehrere Studien an Patienten mit systemischer Sklerose haben gezeigt, dass Patienten mit 24 mmHg ≥ mPAP ≥ 21 mmHg nicht nur eine höhere Sterblichkeit haben, sondern mit größerer Wahrscheinlichkeit eine pulmonale Hypertonie (mPAP ≥ 25 mmHg) entwickeln (
3
). Insgesamt entwickelten 5 – 12 % der Patienten mit linksseitiger Herzerkrankung (PH-LHD) eine Cpc-PH. EU-US- und US-Kohortenstudien zufolge entwickelten 1,5 bis 3 % der Patienten mit Herzinsuffizienz mit reduzierter Ejektionsfraktion (HFrEF) auch eine Cpc-PH; und zwischen 2 und 5 % der Patienten mit Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) entwickelten eine Cpc-PH (aufgrund einer Lungengefäßerkrankung und des allgemeinen metabolischen Syndroms). Pulmonale Hypertonie ist mit einer zweifach erhöhten Mortalität bei Menschen mit HFrEF und HFpEF verbunden. Die 3- und 5-Jahres-Sterblichkeitsraten für PH-LHD lagen bei 42,3 bzw. 52,6 % in einer Kohortenstudie mit 4.621 Patienten mit dieser Erkrankung (
8
). Alle diese klinischen Kohortenstudien zu PH-LHD stammen jedoch aus westlichen Ländern. Die jährliche Konversionsrate von Ipc-PH zu Cpc-PH beträgt etwa 3 bis 6 % (
9
). Bei bestimmten Personengruppen mit systemischer Sklerose können eine Linksherzerkrankung und eine PAH gleichzeitig vorliegen, was das Risiko einer Cpc-PH erhöht. Ältere Menschen, Diabetiker (30–40 %) und Patienten mit chronischer Nierenerkrankung (GFR < 60 ml/min/1,73 m 2 ) haben häufiger einen gemischten pH-Wert (
10
).In China fehlen derzeit repräsentative Daten zu PH-LHD, insbesondere in Form von Langzeit-Follow-up-Berichten. Daher sind multizentrische Registrierungsstudien und die Erstellung einer PH-LHD/Cpc-PH-Datenbank auf der Grundlage von RHC als Standarddiagnose in China dringend erforderlich.Pathophysiologische MechanismenCpc-PH beinhaltet komplexe pathophysiologische Mechanismen. Der Anstieg des PAWP spiegelt eine Funktionsstörung des linken Vorhofs wider. Dieser Druckanstieg überträgt sich auf den Lungenkreislauf und führt zu einem Anstieg von PAP und PVR sowie einer Abnahme der Pulmonalarterien-Compliance (PAC). Dies führt schließlich zu einer endothelialen Dysfunktion, einem pulmonalvaskulären Umbau und einer weiteren Erhöhung der Nachlast des rechten Ventrikels.Mechanismen der pulmonalen Gefäßumgestaltung und FibroseDas zentrale pathologische Merkmal der Cpc-PH ist der Umbau der Lungengefäße. Er ist gekennzeichnet durch Intimaproliferation, Mediahypertrophie und Adventitiafibrose der Lungenarteriolen. In den frühen Stadien führt ein erhöhter Füllungsdruck im linken Ventrikel zu einer Beeinträchtigung des pulmonalvenösen Rückflusses und einem erhöhten Lungengefäßdruck, jedoch zu keinen signifikanten strukturellen Veränderungen. Diese Erkrankung wird als IPC-PH (passive PH) bezeichnet (
11
). Im weiteren Krankheitsverlauf kommt es zu strukturellen und funktionellen Veränderungen der Lungenarterien mit Verdickung und Verhärtung der Gefäßwände, Einengung des Lumens, erhöhtem Widerstand und dauerhaft erhöhtem Lungengefäßwiderstand, was zu Rechtsherzinsuffizienz und dem Eintritt in die Cpc-PH-Phase führt (
12
). Bei Patienten mit Cpc-PH kommt es häufiger zu Funktionsstörungen des rechten Ventrikels und zu einem Umbau der Pulmonalarterie, mit signifikanten Merkmalen eines hypertrophen Umbaus der distalen Pulmonalarterie, einer Fibrose und eines Lumenverschlusses (
13
,
14
).Makrophagen interagieren mit pulmonalvaskulären Endothelzellen und treiben so den pulmonalvaskulären Umbau voran. Dabei kommt es zu einer Zunahme von CD68 + -Zellen (Makrophagen-Subtyp M2) in der Adventitiaschicht. Adventitiafibroblasten könnten die primären Mediatoren ihrer Aktivierung und Polarisierung sein. Das Fettsäure-bindende Protein 5 (FABP5) verschlimmert die pulmonalarterielle Fibrose durch Aktivierung des Wnt/ β -Catenin-Signalwegs, der in Cpc-PH-Mausmodellen signifikant hochreguliert ist und eng mit der pulmonalarteriellen Fibrose verwandt ist. In-vitro- Experimente haben gezeigt, dass die Hemmung von FABP5 (mittels siRNA oder Antagonisten) TGF-β1-induzierte fibrotische Reaktionen abschwächen kann. Spezifische alveoläre Kapillarendothelzellen (Plvap + ) können das mesenchymale Gen aSMA vorübergehend aktivieren und so die Umgestaltung der extrazellulären Matrix, die Gefäßintegrität und die Zell-Zell-Interaktionen beeinflussen (
15
,
16
).SBFI-26 ist ein selektiver FABP5-Inhibitor, der Fibrose durch Modulation des Wnt/ β -Catenin-Signalwegs hemmen kann. Dadurch werden Fibrose und Remodellierung der Lungenarterie in Tiermodellen signifikant unterdrückt und die Wanddicke der Lungenarterie sowie die Kollagenablagerung reduziert. Die zirkuläre RNA circALMS1 ist bei Patienten mit pulmonaler arterieller Hypertonie herunterreguliert, und ihre Überexpression kann die Proliferation und Migration pulmonaler mikrovaskulärer Endothelzellen durch Unterdrückung des miR-17-3p/YTHDF2-Signalwegs hemmen und so die Rechtsherzfunktion verbessern (
17
,
18
).Neurohumorale MechanismenDazu gehören sympathische Überaktivierung, Aktivierung des Renin-Angiotensin-Aldosteron-Systems (RAAS), entzündliche Zellinfiltration und Reaktionen auf oxidativen Stress. Angiotensin II (Ang II) aktiviert den RhoA/Rho-Kinase-Signalweg, hemmt die Aktivität der Myosin-Leichtkettenphosphatase (MLCP) und der endothelialen Stickoxidsynthase (eNOS), was zu Vasokonstriktion und endothelialer Dysfunktion führt (
19
). Die ACE2-Ang (
1
–
7
)-Mas-Achse fördert die Freisetzung von Stickoxid (NO) und Prostaglandinen und übt vasodilatatorische, antiproliferative und entzündungshemmende Effekte aus, wobei eine erhöhte Aktivität die pulmonale Hämodynamik verbessert (
20
). Rekombinantes humanes lösliches ACE2 (rhACE2) kann den pulmonalarteriellen Druck verbessern und oxidativen Stress reduzieren (
21
,
22
).Funktionsstörung des linken VorhofsHFpEF, HFrEF und VHD können im Rahmen des Umbauprozesses zu erhöhtem Druck im linken Vorhof (LAP), erhöhtem Volumen und anschließender Vergrößerung des linken Vorhofs, beeinträchtigter Kontraktilität und interstitieller Fibrose führen (
23
). Der Umbau des linken Vorhofs schwächt dessen Barrierefunktion, wodurch passiv Druck auf die Lungengefäße übertragen wird, was zu erhöhtem Lungenvenendruck und Lungenstauung führt (
24
). Plötzliche Erhöhungen des LAP können zu einem „alveolarkapillären Spannungsversagen“ führen, wodurch die alveolarkapilläre Barriere zerstört wird und ein Lungenödem verursacht wird (
25
). Unter dem Einfluss einer endothelialen Dysfunktion, neurohumoraler Faktoren und einer Infiltration entzündlicher Zellen führen anhaltende Veränderungen des LAP zu strukturellen Anomalien der Lungengefäße und einem erhöhten pulmonalvaskulären Widerstand (
26
–
28
).Pulmonale vaskuläre endotheliale DysfunktionEine Schädigung der Endothelzellen führt zu Vasokonstriktion, Entzündungsreaktionen und Fibrose. Vasoreaktive Substanzen wie Stickstoffmonoxid (NO), Prostacyclin (PGI2) und Endothelin-1 (ET-1) regulieren gemeinsam die pulmonale Vasodilatation und -konstriktion (
29
). NO wird von eNOS synthetisiert und freigesetzt, wodurch die Blutgefäße über den NO-sGC-cGMP-PKG-Signalweg erweitert, die Proliferation glatter Muskelzellen gehemmt und der pulmonale Gefäßumbau verlangsamt wird (
30
). Erhöhte Aldosteronspiegel können oxidativen Stress auslösen, die ET-B-Rezeptorsignalisierung beeinträchtigen, die NO-Synthese und Bioverfügbarkeit verringern. Gleichzeitig kann eine übermäßige ET-1-Produktion die eNOS-Expression hemmen und so die NO-Sekretion verringern (
31
,
32
).Entzündungs- und ChemotaxismechanismenCXCL8 (IL-") und seine Rezeptoren CXCR1 und CXCR2 sind bei pulmonaler arterieller Hypertonie hochreguliert, wobei CXCL8 durch Bindung an CXCR1/2 die Rekrutierung und Aktivierung von Entzündungszellen fördert und so die Entzündung und den Umbau der Pulmonalarterie verschlimmert. Die Interaktion zwischen CXCL10 und seinem Rezeptor CXCR3 spielt eine bedeutende Rolle in der Pathogenese der PH, wobei CXCL10 die Chemotaxis von T-Zellen und natürlichen Killerzellen induziert und an der Entzündungsreaktion der Pulmonalarterie beteiligt ist. Die Signalwege von CXCL12 und seinen Rezeptoren CXCR4 und ACKR3 spielen ebenfalls wichtige Rollen bei PH, wobei die CXCL12-CXCR4-Achse nicht nur die Migration von Entzündungszellen reguliert, sondern auch die Proliferation von PASMCs über den PI3K/Akt-Signalweg fördert (
33
–
36
).Genetische Faktoren und molekulare SignalwegmechanismenBMPR-II ist ein Mitglied der TGF- β- Superfamilie. Heterozygote Mutationen finden sich bei 80 % der Patienten mit familiärer PH und 20 % der Patienten mit sporadischer PH, was es zu einem pathogenen Schlüsselgen für primäre und hereditäre PH macht (
37
–
39
). Es beeinflusst Zellproliferation, Differenzierung, Gewebereparatur, Entzündung und Angiogenese über Smad1/5/8 und nicht-Smad-Signalwege. Die Mutationsrate des BMP9-Gens beträgt in ostasiatischen Populationen bis zu 6,7 %. Die Mutationen reduzieren den zirkulierenden BMP9-Spiegel, schwächen die vaskulären Fähigkeiten zur Apoptose und Schädigung und erhöhen das Risiko für idiopathische PH um mehr als das 22-Fache (
40
–
42
). Der PPARγ-p53-Signalweg reguliert Zellproliferation, Apoptose und Entzündungsreaktionen und beeinflusst die pulmonale Gefäßumgestaltung (
43
–
47
). Auch andere Genmutationen (wie SMAD1, SMAD4, SMAD9, CAV1 und KCNK3) wurden in BMPR-II-Mutations-negativen Familien identifiziert (
48
–
50
). Unter Stressbedingungen wird CLIC4 stark exprimiert, was die Internalisierung und den Abbau von BMPR-II fördert, den BMP-Signalweg hemmt und eine phänotypische Verschiebung in Endothelzellen in Richtung antiapoptotischer und proliferativer Zustände induziert (
51
–
57
).Bei der eingehenden Untersuchung der Pathogenese von Cpc-PH deuten immer mehr Hinweise darauf hin, dass Mutationen in mehreren funktionellen Genen (wie BMPRII, ALK1, CAV1, TBX4, KCNK3, SMAD8, EIF2AK4 usw.) eine wichtige Rolle bei der Pathogenese der Krankheit spielen. Derzeit gehen Forscher im Allgemeinen davon aus, dass das Auftreten von PH ähnlich wie bei Tumoren verläuft, mit einem „Sekundärschlag“-Mechanismus, bei dem genetische und Umweltfaktoren gemeinsam das Auftreten und die Entwicklung der Krankheit fördern.Gentherapie und molekulare SignalwegmechanismenExperimente mit p53-Nanopartikeln und AAV-Vektor-vermittelter Freisetzung normaler BMPR2-Gene haben vielversprechende Ergebnisse gezeigt. Lipid-Nanopartikel (LNPs), die CRISPR-Cas9-Systeme freisetzen, können Genmutationen in PASMCs reparieren (
58
–
60
). Canagliflozin lindert PH-Symptome durch Aktivierung von PPARγ und Hemmung seiner S225-Phosphorylierung und zeigt in Tiermodellen gute Schutzeffekte. Die Aufrechterhaltung der pulmonalvaskulären Homöostase durch Deubiquitinierung zur Aktivierung der ALK2-Smad1/5/9-PPARγ-Achse mit Hochregulierung von BRCC3 kann PH-Symptome lindern (
61
–
64
) (
Abbildung 2
). Abbildung 2
und seine Rezeptoren CXCR1 und CXCR2 sind bei pulmonaler arterieller Hypertonie hochreguliert, wobei CXCL8 durch Bindung an CXCR1/2 die Rekrutierung und Aktivierung von Entzündungszellen fördert und so die Entzündung und den Umbau der Pulmonalarterie verschlimmert. Die Interaktion zwischen CXCL10 und seinem Rezeptor CXCR3 spielt eine bedeutende Rolle in der Pathogenese der PH, wobei CXCL10 die Chemotaxis von T-Zellen und natürlichen Killerzellen induziert und an der Entzündungsreaktion der Pulmonalarterie beteiligt ist. Die Signalwege von CXCL12 und seinen Rezeptoren CXCR4 und ACKR3 spielen ebenfalls wichtige Rollen bei PH, wobei die CXCL12-CXCR4-Achse nicht nur die Migration von Entzündungszellen reguliert, sondern auch die Proliferation von PASMCs über den PI3K/Akt-Signalweg fördert (
33
–
36
).Genetische Faktoren und molekulare SignalwegmechanismenBMPR-II ist ein Mitglied der TGF- β- Superfamilie. Heterozygote Mutationen finden sich bei 80 % der Patienten mit familiärer PH und 20 % der Patienten mit sporadischer PH, was es zu einem pathogenen Schlüsselgen für primäre und hereditäre PH macht (
37
–
39
). Es beeinflusst Zellproliferation, Differenzierung, Gewebereparatur, Entzündung und Angiogenese über Smad1/5/8 und nicht-Smad-Signalwege. Die Mutationsrate des BMP9-Gens beträgt in ostasiatischen Populationen bis zu 6,7 %. Die Mutationen reduzieren den zirkulierenden BMP9-Spiegel, schwächen die vaskulären Fähigkeiten zur Apoptose und Schädigung und erhöhen das Risiko für idiopathische PH um mehr als das 22-Fache (
40
–
42
). Der PPARγ-p53-Signalweg reguliert Zellproliferation, Apoptose und Entzündungsreaktionen und beeinflusst die pulmonale Gefäßumgestaltung (
43
–
47
). Auch andere Genmutationen (wie SMAD1, SMAD4, SMAD9, CAV1 und KCNK3) wurden in BMPR-II-Mutations-negativen Familien identifiziert (
48
–
50
). Unter Stressbedingungen wird CLIC4 stark exprimiert, was die Internalisierung und den Abbau von BMPR-II fördert, den BMP-Signalweg hemmt und eine phänotypische Verschiebung in Endothelzellen in Richtung antiapoptotischer und proliferativer Zustände induziert (
51
–
57
).Bei der eingehenden Untersuchung der Pathogenese von Cpc-PH deuten immer mehr Hinweise darauf hin, dass Mutationen in mehreren funktionellen Genen (wie BMPRII, ALK1, CAV1, TBX4, KCNK3, SMAD8, EIF2AK4 usw.) eine wichtige Rolle bei der Pathogenese der Krankheit spielen. Derzeit gehen Forscher im Allgemeinen davon aus, dass das Auftreten von PH ähnlich wie bei Tumoren verläuft, mit einem „Sekundärschlag“-Mechanismus, bei dem genetische und Umweltfaktoren gemeinsam das Auftreten und die Entwicklung der Krankheit fördern.Gentherapie und molekulare SignalwegmechanismenExperimente mit p53-Nanopartikeln und AAV-Vektor-vermittelter Freisetzung normaler BMPR2-Gene haben vielversprechende Ergebnisse gezeigt. Lipid-Nanopartikel (LNPs), die CRISPR-Cas9-Systeme freisetzen, können Genmutationen in PASMCs reparieren (
58
–
60
). Canagliflozin lindert PH-Symptome durch Aktivierung von PPARγ und Hemmung seiner S225-Phosphorylierung und zeigt in Tiermodellen gute Schutzeffekte. Die Aufrechterhaltung der pulmonalvaskulären Homöostase durch Deubiquitinierung zur Aktivierung der ALK2-Smad1/5/9-PPARγ-Achse mit Hochregulierung von BRCC3 kann PH-Symptome lindern (
61
–
64
) (
Abbildung 2
). Abbildung 2
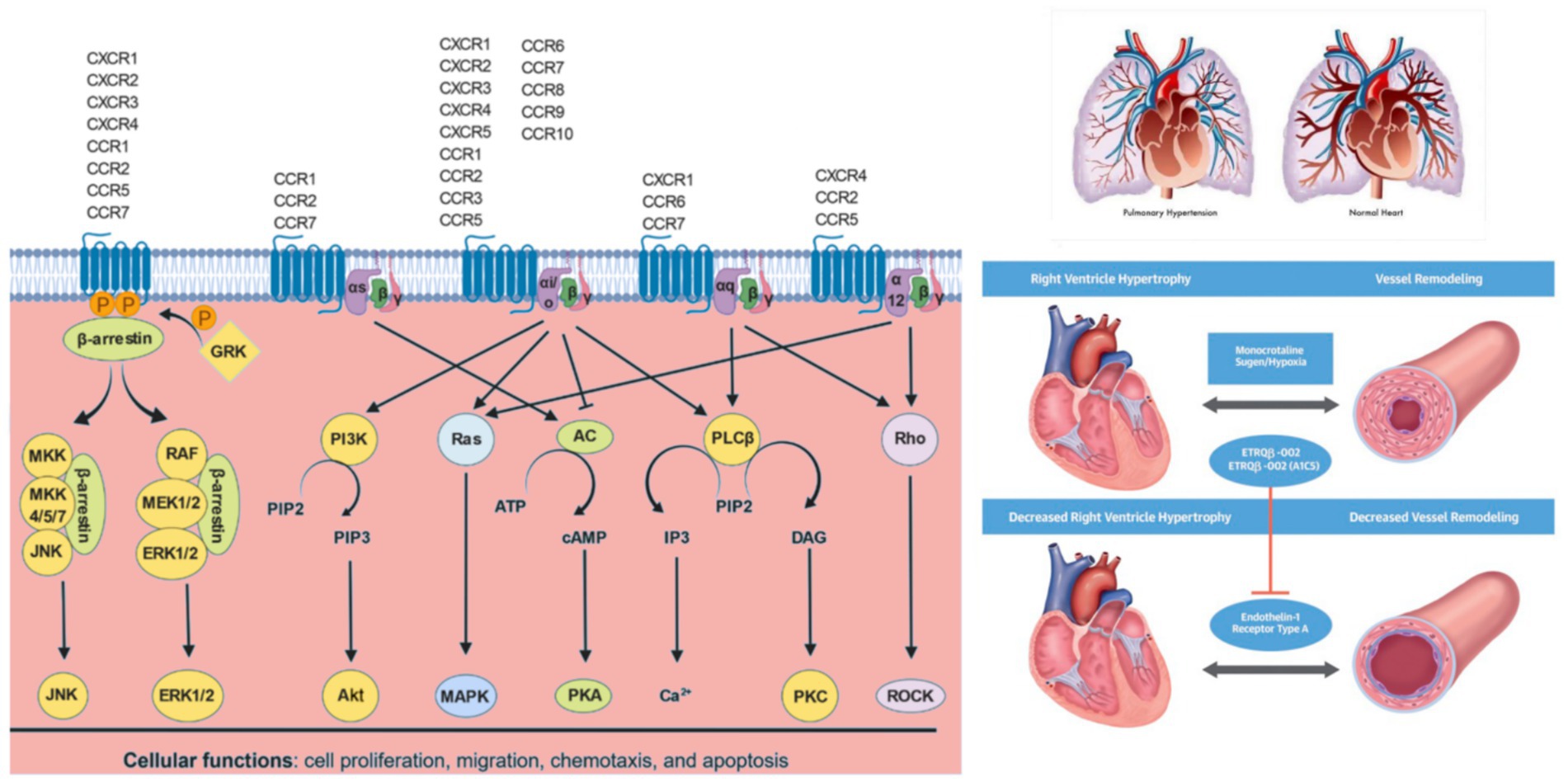 Abbildung 2. Chemokin-Signalwege: Chemokine binden an ihre verwandten Chemokinrezeptoren, die auf verschiedenen Zelltypen exprimiert werden. Bei Rezeptoraktivierung dissoziiert das G-Protein in Gα- und Gβγ - Untereinheiten. Abhängig von der/den spezifischen Gα-Untereinheit(en), an die der Chemokinrezeptor gekoppelt ist, werden verschiedene nachgeschaltete Signalwege reguliert. Das Gαs-Protein aktiviert die Adenylatcyclase (AC) zur Produktion von cAMP, das wiederum die cAMP-abhängige PKA aktiviert. Gαi/o inaktiviert AC und begrenzt so die cAMP-Spiegel und die PKA-Aktivität. Gαi/o-Proteine aktivieren PI3Ks, das Phosphatidylinositol-4,5-bisphosphat (PIP2) in Phosphatidylinositol(3,4,5)-trisphosphat (PIP3) umwandelt, das wiederum PKB (Akt) aktiviert. Gαi/o- und Gα12-Proteine aktivieren Ras, das wiederum verschiedene MAPKs aktiviert. Gαi/o- und Gαq-Proteine aktivieren PLCβ, das PIP2 zu IP3 und DAG katalysiert. IP3 reguliert außerdem die intrazellulären freien Calciumwerte (Ca 2+ ), während DAG PKC aktiviert. Gαq- und Gα12-Proteine aktivieren auch GTPasen der Rho-Familie (Rho), die wiederum Rho-assoziierte PK (ROCK) aktivieren. GPCR-Kinasen (GRK) phosphorylieren GPCRs, wodurch β-Arrestine an GPCRs binden und sie internalisieren können, was zu einem Rezeptor-Recycling führen kann, wenn die Rezeptorphosphorylierung durch Proteinphosphatase 2 (PP2A) oder Abbau in Lysosomen umgekehrt wird. An GPCRs gebundenes β-Arrestin kann auch MAPK-Signalwege wie JNK und ERKs 1 und 2 (ERK1/2) aktivieren. Diagnostische MethodenDie Diagnose der Cpc-PH basiert auf einer Kombination hämodynamischer, radiologischer und klinischer Befunde. Die Rechtsherzkatheterisierung (RHC) ist nach wie vor der Goldstandard zur Unterscheidung von PH-LHD/Cpc-PH von anderen Formen der PH. Zur hämodynamischen Klassifizierung wurden mPAP, PAWP, PVR und DPG direkt gemessen. Diastolischer Druckgradient (DPG) = pulmonaler diastolischer Blutdruck-PAWP, DPG ≥ 7 mmHg weist auf eine gemischte pulmonale Hypertonie hin; transpulmonaler Druckgradient (TPG) = mPAP-PAWP, TPG ≥ 12 mmHg kann auf eine gemischte Form hinweisen. Immer mehr Daten deuten darauf hin, dass das Verhältnis von Schlagvolumen zu pulmonalarteriellem Pulsdruck, ein Ersatzmaß für die pulmonalarterielle Compliance (PAC), mit schlechten Ergebnissen bei Patienten mit Herzinsuffizienz verbunden ist (
65
). Dies legt nahe, dass PACs bei der Beurteilung früher pulmonaler Gefäßerkrankungen größere Aufmerksamkeit geschenkt werden sollten (
66
) (
Abbildung 3
). Abbildung 3
Abbildung 2. Chemokin-Signalwege: Chemokine binden an ihre verwandten Chemokinrezeptoren, die auf verschiedenen Zelltypen exprimiert werden. Bei Rezeptoraktivierung dissoziiert das G-Protein in Gα- und Gβγ - Untereinheiten. Abhängig von der/den spezifischen Gα-Untereinheit(en), an die der Chemokinrezeptor gekoppelt ist, werden verschiedene nachgeschaltete Signalwege reguliert. Das Gαs-Protein aktiviert die Adenylatcyclase (AC) zur Produktion von cAMP, das wiederum die cAMP-abhängige PKA aktiviert. Gαi/o inaktiviert AC und begrenzt so die cAMP-Spiegel und die PKA-Aktivität. Gαi/o-Proteine aktivieren PI3Ks, das Phosphatidylinositol-4,5-bisphosphat (PIP2) in Phosphatidylinositol(3,4,5)-trisphosphat (PIP3) umwandelt, das wiederum PKB (Akt) aktiviert. Gαi/o- und Gα12-Proteine aktivieren Ras, das wiederum verschiedene MAPKs aktiviert. Gαi/o- und Gαq-Proteine aktivieren PLCβ, das PIP2 zu IP3 und DAG katalysiert. IP3 reguliert außerdem die intrazellulären freien Calciumwerte (Ca 2+ ), während DAG PKC aktiviert. Gαq- und Gα12-Proteine aktivieren auch GTPasen der Rho-Familie (Rho), die wiederum Rho-assoziierte PK (ROCK) aktivieren. GPCR-Kinasen (GRK) phosphorylieren GPCRs, wodurch β-Arrestine an GPCRs binden und sie internalisieren können, was zu einem Rezeptor-Recycling führen kann, wenn die Rezeptorphosphorylierung durch Proteinphosphatase 2 (PP2A) oder Abbau in Lysosomen umgekehrt wird. An GPCRs gebundenes β-Arrestin kann auch MAPK-Signalwege wie JNK und ERKs 1 und 2 (ERK1/2) aktivieren. Diagnostische MethodenDie Diagnose der Cpc-PH basiert auf einer Kombination hämodynamischer, radiologischer und klinischer Befunde. Die Rechtsherzkatheterisierung (RHC) ist nach wie vor der Goldstandard zur Unterscheidung von PH-LHD/Cpc-PH von anderen Formen der PH. Zur hämodynamischen Klassifizierung wurden mPAP, PAWP, PVR und DPG direkt gemessen. Diastolischer Druckgradient (DPG) = pulmonaler diastolischer Blutdruck-PAWP, DPG ≥ 7 mmHg weist auf eine gemischte pulmonale Hypertonie hin; transpulmonaler Druckgradient (TPG) = mPAP-PAWP, TPG ≥ 12 mmHg kann auf eine gemischte Form hinweisen. Immer mehr Daten deuten darauf hin, dass das Verhältnis von Schlagvolumen zu pulmonalarteriellem Pulsdruck, ein Ersatzmaß für die pulmonalarterielle Compliance (PAC), mit schlechten Ergebnissen bei Patienten mit Herzinsuffizienz verbunden ist (
65
). Dies legt nahe, dass PACs bei der Beurteilung früher pulmonaler Gefäßerkrankungen größere Aufmerksamkeit geschenkt werden sollten (
66
) (
Abbildung 3
). Abbildung 3
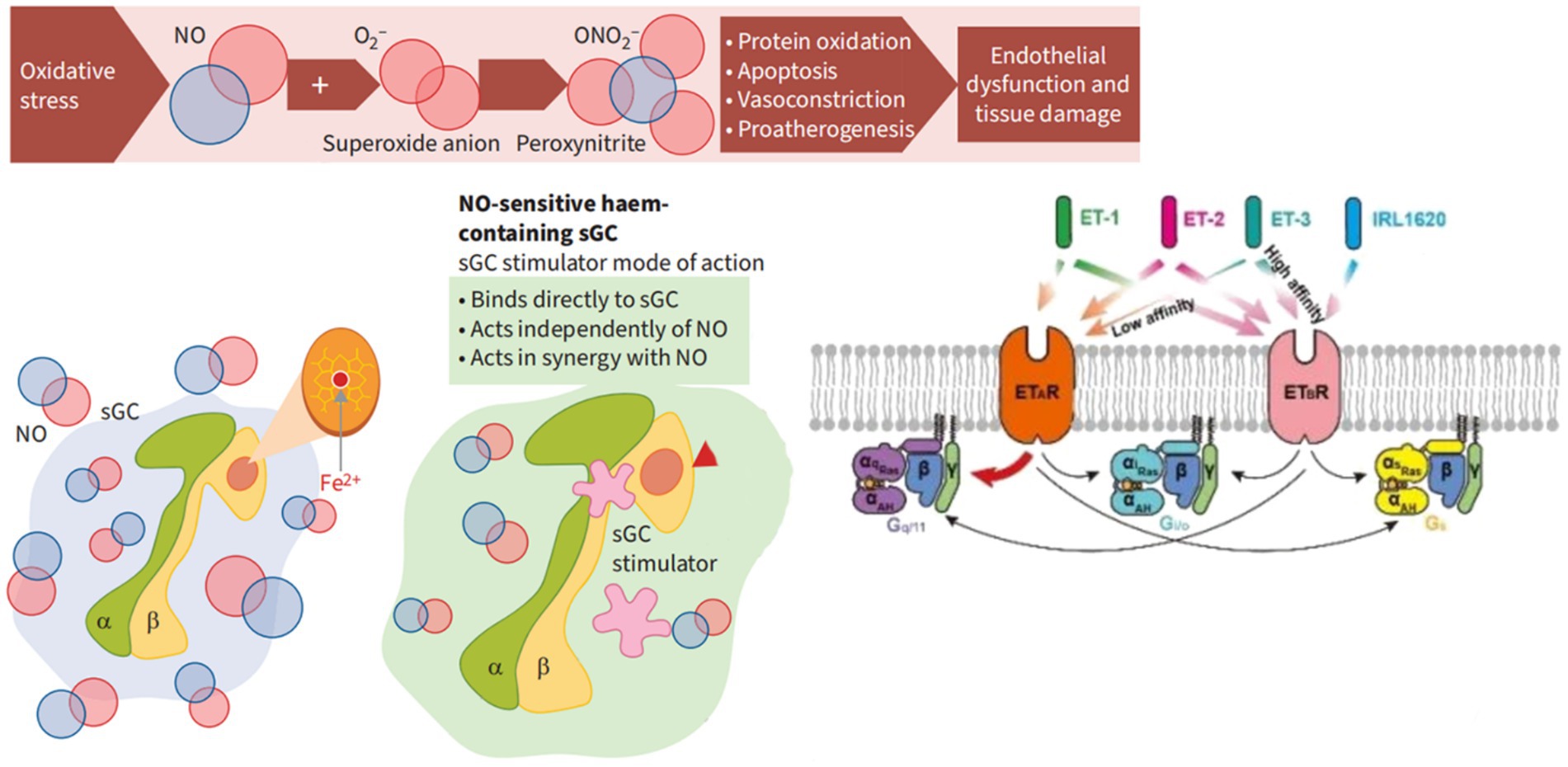 Abbildung 3. Der Wirkungsmechanismus von Stimulanzien und Aktivatoren der löslichen Guanylatcyclase (sGC). α, SGC-α-Untereinheit; β-Wert, sGC-β-Untereinheit; cGMP, zyklisches Guanosinmonophosphat; Fe2 + , Eisenionen oxidieren in einem +2-Zustand; NO, Stickstoffmonoxid; O2- , Superoxidanion; ONO2- , Peroxynitrit. Obwohl die Rechtsherzkatheterisierung der Goldstandard zur Diagnose von PH ist, ist sie invasiv. Zukünftige Forschung wird sich auf die Entwicklung präziserer nichtinvasiver Diagnosetechniken konzentrieren, wie etwa die Verwendung moderner bildgebender Verfahren (z. B. Elektrokardiogramm, Magnetresonanztomographie, Computertomographie) und Biomarkertests, um den hämodynamischen Status und die pulmonalvaskuläre Architektur von Patienten genauer beurteilen zu können (
67
). Die transthorakale Echokardiographie (TTE) ist das wichtigste Screening-Element zur Messung des Schweregrads von PH. Mit der TTE werden eine Dilatation des rechten Ventrikels, das Vorhandensein einer Klappeninsuffizienz und das Vorhandensein einer linksventrikulären systolischen oder diastolischen Dysfunktion festgestellt (
68
). Ventilations-/Perfusionsscans werden auch empfohlen, wenn eine Herzinsuffizienz (HF) mit Hyperkoagulabilität einhergeht. Ventilations-/Perfusionsscans bleiben im klinischen Arbeitsablauf unverzichtbar, da eine chronische thromboembolische pulmonale Hypertonie bei normaler Perfusion unwahrscheinlich ist (
69
). Röntgenaufnahmen des Brustkorbs können eine Kardiomegalie und erweiterte Pulmonalarterien sowie Lungenparenchym- oder Brustwandanomalien zeigen. Eine CT-Angiographie des Brustkorbs gilt zwar als weniger sensitiv als die Ventilations-Perfusions-Szintigraphie, kann aber Anzeichen einer chronischen thromboembolischen Erkrankung wie Füllungsdefekte oder keilförmige oder unregelmäßige lineare Trübungen aufgrund einer früheren Thrombose offenbaren. Sie wird verwendet, um das Vorhandensein einer Pulmonalarterienstenose oder okklusiver Läsionen festzustellen (
70
). Serumspiegel des Amino-terminalen natriuretischen Peptids (BNP) können in die Risikostratifizierung einbezogen werden, da sie stark mit dem Schweregrad der pulmonalen Hypertonie korrelieren und zur Vorhersage des Überlebens verwendet werden können (
71
). Der 6MWT spiegelt die allgemeine Belastungstoleranz und die Fähigkeit zu täglicher Aktivität wider und eignet sich zur Beurteilung des Funktionsstatus und des Rehabilitationseffekts. BNP quantifiziert die kardiale Belastung und den Grad der Verletzung und eignet sich zur Beurteilung des pathophysiologischen Schweregrads und der Wirksamkeit der Behandlung. Die Kombination beider Verfahren kann die umfassendere Untersuchung von Patienten mit Cpc-PH verbessern. Bei Patienten mit PH spiegelt ein erhöhter BNP-Wert eine Drucküberlastung des rechten Ventrikels und eine fortschreitende Rechtsherzinsuffizienz wider (
72
,
73
).CardioMEMS ist ein implantierbares Drucküberwachungssystem, das über einen Katheter in die Pulmonalarterie eingesetzt wird, um PAP, CO, mPAP, PAWP und andere Werte in Echtzeit zu überwachen und so die Behandlung von Herzinsuffizienz und PH zu steuern (
74
). Der Arzt kann den Zustand des Patienten aus der Ferne überwachen und den Behandlungsplan umgehend anpassen, da die Daten drahtlos an ein externes Gerät übertragen werden. Dies ist für die Beurteilung des Zustands von entscheidender Bedeutung. In einer Studie erfuhren Patienten, die mit CardioMEMS behandelt wurden, eine signifikante Verringerung der Krankenhausaufenthalte und eine deutliche Verbesserung ihrer Lebensqualität. Die CHAMPION-Studie ist eine multizentrische, randomisierte, kontrollierte Studie zur Bewertung von CardioMEMS bei Patienten mit Herzinsuffizienz (
75
). Die Ergebnisse zeigten, dass Patienten, die CardioMEMS verwendeten, innerhalb von 6 Monaten eine signifikante Verringerung der Krankenhausaufenthalte und eine deutliche Verbesserung ihrer Lebensqualität erfuhren. In der PULSAR-Studie wurde der Einsatz von CardioMEMS bei Patienten mit pulmonaler Hypertonie weiter untersucht. Es wurde festgestellt, dass CardioMEMS die 6-Minuten-Gehstrecke (6MWD) und die NT-proBNP-Werte signifikant verbesserte (
76
). In einer Langzeit-Nachuntersuchung zeigten mit CardioMEMS behandelte Patienten über einen Zeitraum von zwei Jahren eine anhaltende Verbesserung des pulmonalarteriellen Drucks und der Herzfunktion, ohne dass gerätebedingte schwerwiegende Nebenwirkungen auftraten (
77
).In einer Studie mit 208 Patienten mit chronischer Herzinsuffizienz war BNP signifikant invers mit der 6MWD verbunden (r = −0,61), und die kombinierte prädiktive Wirksamkeit der beiden Messungen war besser als die einer einzelnen Messung (
78
). Bei Patienten mit PH weisen 6MWD < 300 m und BNP > 300 pg/ml auf ein sehr hohes Risiko hin und erfordern eine intensive Therapie. Studien haben gezeigt, dass sich das Sterberisiko bei Patienten mit PH mit jeder Verringerung der 6MWD um 50 m signifikant erhöht; eine Verringerung der 6MWD um >50 m über 24 Wochen ist mit einer mindestens vierfach erhöhten Mortalität verbunden (
79
). Die 6MWD wird häufig verwendet, um die Wirksamkeit zielgerichteter Medikamente (z. B. Sotatercept), chirurgischer Eingriffe oder Bewegungsrehabilitation zu bewerten. So ging beispielsweise eine durchschnittliche Erhöhung der 6MWD um 40,8 m, gefolgt von einer Pharmakotherapie, mit einer gleichzeitigen Verbesserung des pulmonalvaskulären Widerstands und der BNP-Werte einher (
80
). Eine Verbesserung von 30 bis 50 m im 6MWT gilt als Schwelle für einen klinischen Nutzen bei Patienten mit PH und wird als minimale klinisch bedeutsame Differenz (MCID) bezeichnet (
81
). Die maximale Sauerstoffaufnahme kann im 6MWT nicht direkt gemessen werden und die Ergebnisse werden leicht von Faktoren wie Alter, Geschlecht und Korridorlänge beeinflusst, sodass sie in Kombination mit anderen Indikatoren umfassend beurteilt werden muss. Es wurde gezeigt, dass die BNP-Werte mit der NYHA-Herzfunktionsklasse signifikant ansteigen (Grad I: 186 ± 22 pg/ml; Grad IV: 266 ± 165 Pikogramm/ml) (
82
). Die ROC-Kurvenanalyse zeigte, dass BNP unerwünschte Ereignisse bei Patienten mit chronischer Herzinsuffizienz mit einer Fläche unter der Kurve von 0,914 (Sensitivität 0,778, Spezifität 0,977) vorhersagt (
83
,
84
).Aktuelle medizinische und chirurgische BehandlungenAuf Grundlage vorhandener Evidenz zeigt die zielgerichtete Therapie von PH-LHD keine signifikante Wirkung bei Patienten mit Cpc-PH. Konventionelle Behandlungsmodalitäten umfassen die Verbesserung der Kontrolle der Grunderkrankungen, eine gezielte medikamentöse Therapie und chirurgische Eingriffe. Die Komplexität der Behandlung erfordert eine präzise hämodynamische Beurteilung und Risikostratifizierung, wobei die potenziellen Risiken zielgerichteter Medikamente zu beachten sind. Es wird empfohlen, Medikamente der ersten Wahl, darunter Betablocker, RAAS-Hemmer, Stimulanzien der löslichen Guanylatcyclase und Natrium-Glucose-Cotransporter-2-Hemmer, in einer sinnvollen Kombination einzusetzen, um die Symptome der Patienten zu lindern, das Fortschreiten der Krankheit zu verzögern und die Sterblichkeit zu senken. Cpc-PH-Patienten mit metabolischem Syndrom und anderen Erkrankungen sollten ebenfalls behandelt werden. Obwohl es nur begrenzte Evidenz für die gezielte medikamentöse Anwendung von Cpc-PH gibt, sollten zielgerichtete Medikamente in bestimmten Situationen dennoch in Betracht gezogen werden. Die Studienergebnisse mehrerer Medikamentenklassen, die zur Behandlung von Cpc-PH eingesetzt werden können, werden in den folgenden Abschnitten besprochen (
Tabellen 1
–
3
). Bei Ipc-PH kann eine zielgerichtete medikamentöse Therapie schädlich sein, da der pulmonalarterielle Druckanstieg passiv ist und noch nicht zum Umbau der pulmonalarteriellen Gefäße gekommen ist. Bei Cpc-PH können zielgerichtete Medikamente zwar für einige Patienten von Nutzen sein, es fehlen jedoch noch immer klare Screening-Kriterien. Zukünftige Forschung sollte sich auf das Screening von PH-LHD-Patienten konzentrieren, die auf zielgerichtete Medikamente ansprechen, insbesondere Cpc-PH-Patienten mit hohem pulmonalarteriellen Druck, selbst nach Verbesserung der Linksherzfunktion (
85
). Tabelle 1
Abbildung 3. Der Wirkungsmechanismus von Stimulanzien und Aktivatoren der löslichen Guanylatcyclase (sGC). α, SGC-α-Untereinheit; β-Wert, sGC-β-Untereinheit; cGMP, zyklisches Guanosinmonophosphat; Fe2 + , Eisenionen oxidieren in einem +2-Zustand; NO, Stickstoffmonoxid; O2- , Superoxidanion; ONO2- , Peroxynitrit. Obwohl die Rechtsherzkatheterisierung der Goldstandard zur Diagnose von PH ist, ist sie invasiv. Zukünftige Forschung wird sich auf die Entwicklung präziserer nichtinvasiver Diagnosetechniken konzentrieren, wie etwa die Verwendung moderner bildgebender Verfahren (z. B. Elektrokardiogramm, Magnetresonanztomographie, Computertomographie) und Biomarkertests, um den hämodynamischen Status und die pulmonalvaskuläre Architektur von Patienten genauer beurteilen zu können (
67
). Die transthorakale Echokardiographie (TTE) ist das wichtigste Screening-Element zur Messung des Schweregrads von PH. Mit der TTE werden eine Dilatation des rechten Ventrikels, das Vorhandensein einer Klappeninsuffizienz und das Vorhandensein einer linksventrikulären systolischen oder diastolischen Dysfunktion festgestellt (
68
). Ventilations-/Perfusionsscans werden auch empfohlen, wenn eine Herzinsuffizienz (HF) mit Hyperkoagulabilität einhergeht. Ventilations-/Perfusionsscans bleiben im klinischen Arbeitsablauf unverzichtbar, da eine chronische thromboembolische pulmonale Hypertonie bei normaler Perfusion unwahrscheinlich ist (
69
). Röntgenaufnahmen des Brustkorbs können eine Kardiomegalie und erweiterte Pulmonalarterien sowie Lungenparenchym- oder Brustwandanomalien zeigen. Eine CT-Angiographie des Brustkorbs gilt zwar als weniger sensitiv als die Ventilations-Perfusions-Szintigraphie, kann aber Anzeichen einer chronischen thromboembolischen Erkrankung wie Füllungsdefekte oder keilförmige oder unregelmäßige lineare Trübungen aufgrund einer früheren Thrombose offenbaren. Sie wird verwendet, um das Vorhandensein einer Pulmonalarterienstenose oder okklusiver Läsionen festzustellen (
70
). Serumspiegel des Amino-terminalen natriuretischen Peptids (BNP) können in die Risikostratifizierung einbezogen werden, da sie stark mit dem Schweregrad der pulmonalen Hypertonie korrelieren und zur Vorhersage des Überlebens verwendet werden können (
71
). Der 6MWT spiegelt die allgemeine Belastungstoleranz und die Fähigkeit zu täglicher Aktivität wider und eignet sich zur Beurteilung des Funktionsstatus und des Rehabilitationseffekts. BNP quantifiziert die kardiale Belastung und den Grad der Verletzung und eignet sich zur Beurteilung des pathophysiologischen Schweregrads und der Wirksamkeit der Behandlung. Die Kombination beider Verfahren kann die umfassendere Untersuchung von Patienten mit Cpc-PH verbessern. Bei Patienten mit PH spiegelt ein erhöhter BNP-Wert eine Drucküberlastung des rechten Ventrikels und eine fortschreitende Rechtsherzinsuffizienz wider (
72
,
73
).CardioMEMS ist ein implantierbares Drucküberwachungssystem, das über einen Katheter in die Pulmonalarterie eingesetzt wird, um PAP, CO, mPAP, PAWP und andere Werte in Echtzeit zu überwachen und so die Behandlung von Herzinsuffizienz und PH zu steuern (
74
). Der Arzt kann den Zustand des Patienten aus der Ferne überwachen und den Behandlungsplan umgehend anpassen, da die Daten drahtlos an ein externes Gerät übertragen werden. Dies ist für die Beurteilung des Zustands von entscheidender Bedeutung. In einer Studie erfuhren Patienten, die mit CardioMEMS behandelt wurden, eine signifikante Verringerung der Krankenhausaufenthalte und eine deutliche Verbesserung ihrer Lebensqualität. Die CHAMPION-Studie ist eine multizentrische, randomisierte, kontrollierte Studie zur Bewertung von CardioMEMS bei Patienten mit Herzinsuffizienz (
75
). Die Ergebnisse zeigten, dass Patienten, die CardioMEMS verwendeten, innerhalb von 6 Monaten eine signifikante Verringerung der Krankenhausaufenthalte und eine deutliche Verbesserung ihrer Lebensqualität erfuhren. In der PULSAR-Studie wurde der Einsatz von CardioMEMS bei Patienten mit pulmonaler Hypertonie weiter untersucht. Es wurde festgestellt, dass CardioMEMS die 6-Minuten-Gehstrecke (6MWD) und die NT-proBNP-Werte signifikant verbesserte (
76
). In einer Langzeit-Nachuntersuchung zeigten mit CardioMEMS behandelte Patienten über einen Zeitraum von zwei Jahren eine anhaltende Verbesserung des pulmonalarteriellen Drucks und der Herzfunktion, ohne dass gerätebedingte schwerwiegende Nebenwirkungen auftraten (
77
).In einer Studie mit 208 Patienten mit chronischer Herzinsuffizienz war BNP signifikant invers mit der 6MWD verbunden (r = −0,61), und die kombinierte prädiktive Wirksamkeit der beiden Messungen war besser als die einer einzelnen Messung (
78
). Bei Patienten mit PH weisen 6MWD < 300 m und BNP > 300 pg/ml auf ein sehr hohes Risiko hin und erfordern eine intensive Therapie. Studien haben gezeigt, dass sich das Sterberisiko bei Patienten mit PH mit jeder Verringerung der 6MWD um 50 m signifikant erhöht; eine Verringerung der 6MWD um >50 m über 24 Wochen ist mit einer mindestens vierfach erhöhten Mortalität verbunden (
79
). Die 6MWD wird häufig verwendet, um die Wirksamkeit zielgerichteter Medikamente (z. B. Sotatercept), chirurgischer Eingriffe oder Bewegungsrehabilitation zu bewerten. So ging beispielsweise eine durchschnittliche Erhöhung der 6MWD um 40,8 m, gefolgt von einer Pharmakotherapie, mit einer gleichzeitigen Verbesserung des pulmonalvaskulären Widerstands und der BNP-Werte einher (
80
). Eine Verbesserung von 30 bis 50 m im 6MWT gilt als Schwelle für einen klinischen Nutzen bei Patienten mit PH und wird als minimale klinisch bedeutsame Differenz (MCID) bezeichnet (
81
). Die maximale Sauerstoffaufnahme kann im 6MWT nicht direkt gemessen werden und die Ergebnisse werden leicht von Faktoren wie Alter, Geschlecht und Korridorlänge beeinflusst, sodass sie in Kombination mit anderen Indikatoren umfassend beurteilt werden muss. Es wurde gezeigt, dass die BNP-Werte mit der NYHA-Herzfunktionsklasse signifikant ansteigen (Grad I: 186 ± 22 pg/ml; Grad IV: 266 ± 165 Pikogramm/ml) (
82
). Die ROC-Kurvenanalyse zeigte, dass BNP unerwünschte Ereignisse bei Patienten mit chronischer Herzinsuffizienz mit einer Fläche unter der Kurve von 0,914 (Sensitivität 0,778, Spezifität 0,977) vorhersagt (
83
,
84
).Aktuelle medizinische und chirurgische BehandlungenAuf Grundlage vorhandener Evidenz zeigt die zielgerichtete Therapie von PH-LHD keine signifikante Wirkung bei Patienten mit Cpc-PH. Konventionelle Behandlungsmodalitäten umfassen die Verbesserung der Kontrolle der Grunderkrankungen, eine gezielte medikamentöse Therapie und chirurgische Eingriffe. Die Komplexität der Behandlung erfordert eine präzise hämodynamische Beurteilung und Risikostratifizierung, wobei die potenziellen Risiken zielgerichteter Medikamente zu beachten sind. Es wird empfohlen, Medikamente der ersten Wahl, darunter Betablocker, RAAS-Hemmer, Stimulanzien der löslichen Guanylatcyclase und Natrium-Glucose-Cotransporter-2-Hemmer, in einer sinnvollen Kombination einzusetzen, um die Symptome der Patienten zu lindern, das Fortschreiten der Krankheit zu verzögern und die Sterblichkeit zu senken. Cpc-PH-Patienten mit metabolischem Syndrom und anderen Erkrankungen sollten ebenfalls behandelt werden. Obwohl es nur begrenzte Evidenz für die gezielte medikamentöse Anwendung von Cpc-PH gibt, sollten zielgerichtete Medikamente in bestimmten Situationen dennoch in Betracht gezogen werden. Die Studienergebnisse mehrerer Medikamentenklassen, die zur Behandlung von Cpc-PH eingesetzt werden können, werden in den folgenden Abschnitten besprochen (
Tabellen 1
–
3
). Bei Ipc-PH kann eine zielgerichtete medikamentöse Therapie schädlich sein, da der pulmonalarterielle Druckanstieg passiv ist und noch nicht zum Umbau der pulmonalarteriellen Gefäße gekommen ist. Bei Cpc-PH können zielgerichtete Medikamente zwar für einige Patienten von Nutzen sein, es fehlen jedoch noch immer klare Screening-Kriterien. Zukünftige Forschung sollte sich auf das Screening von PH-LHD-Patienten konzentrieren, die auf zielgerichtete Medikamente ansprechen, insbesondere Cpc-PH-Patienten mit hohem pulmonalarteriellen Druck, selbst nach Verbesserung der Linksherzfunktion (
85
). Tabelle 1
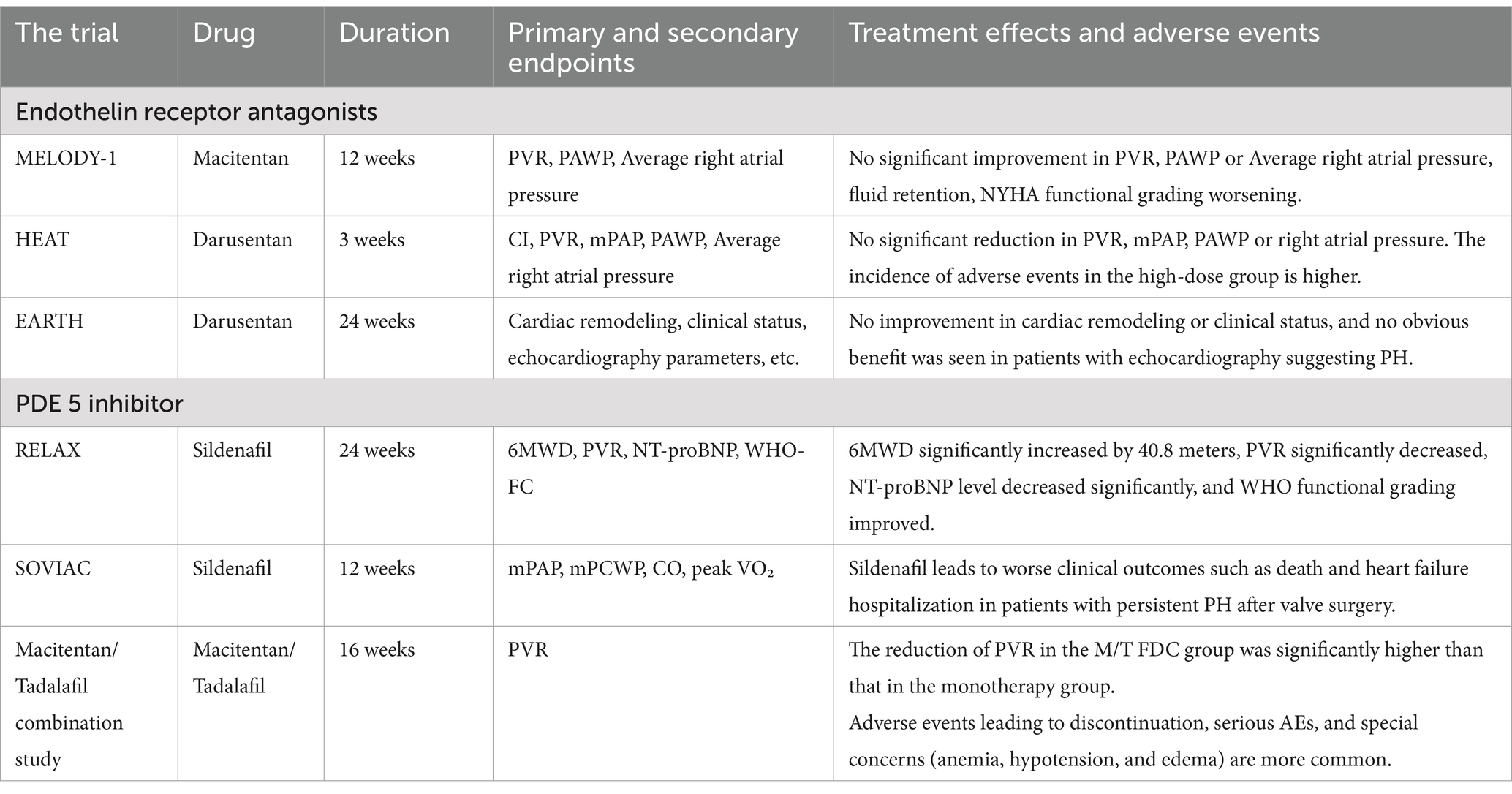 Tabelle 1. Endothelin-Rezeptor-Antagonisten und PDE-5-Hemmer. Tabelle 2
Tabelle 1. Endothelin-Rezeptor-Antagonisten und PDE-5-Hemmer. Tabelle 2
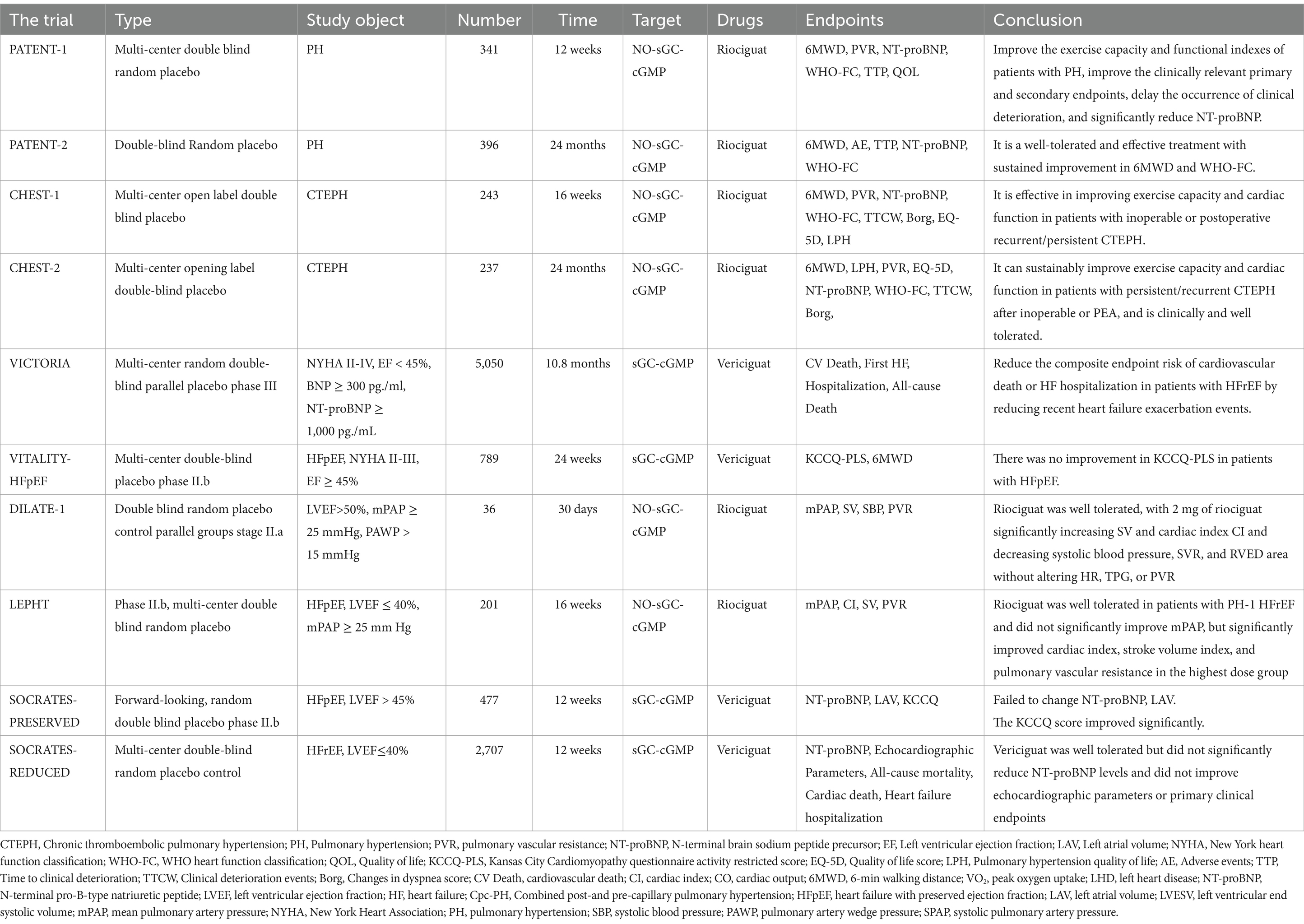 Tabelle 2. Löslicher Guanylatcyclase-Stimulator. Tabelle 3
Tabelle 2. Löslicher Guanylatcyclase-Stimulator. Tabelle 3
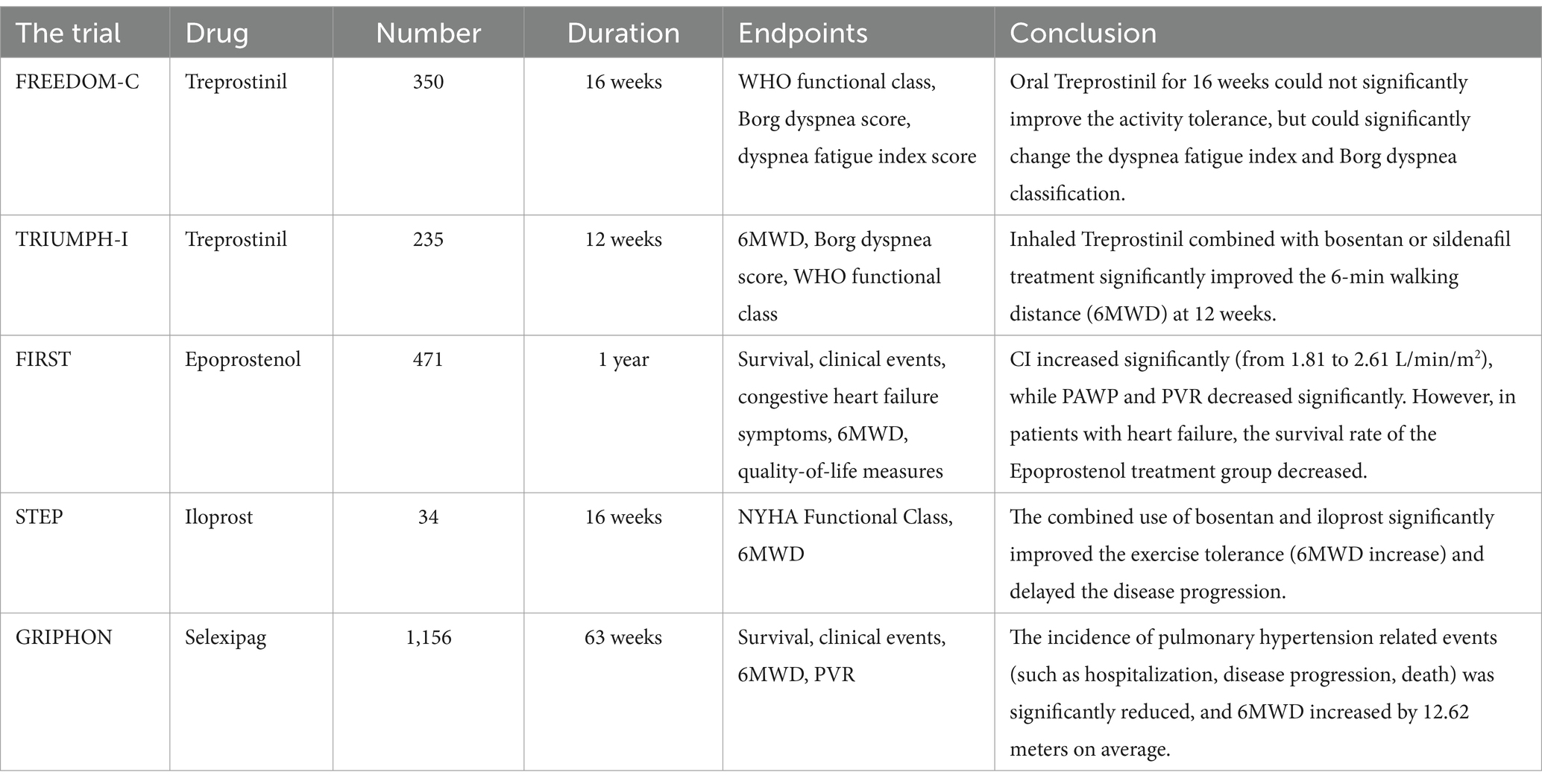 Tabelle 3. PGI2-Medikamente. Endothelin-Rezeptor-AntagonistenBisher wurden vier Arten von ERAs klinisch getestet, nämlich Bosentan, Anlisentan, Masitentan und Sitaxentan. Unter diesen wurde Sitaxentan aufgrund tödlicher Leberschäden weltweit vom Markt genommen (
75
). Endothelin-1 (ET-1) ist an der Gefäßverengung und Zellproliferation beteiligt, indem es Endothelinrezeptoren (ET-A und ET- aktiviert (
86
). Cochrane-Studien haben gezeigt, dass die Verwendung von Endothelinrezeptor-Antagonisten über 3 bis 6 Monate die körperliche Leistungsfähigkeit, die Symptome und die kardiorespiratorischen hämodynamischen Indikatoren der Patienten signifikant verbessert (
87
). Derzeit ist jedoch unklar, ob diese Medikamente die Sterblichkeitsrate der Patienten signifikant senken können (
88
). Bei PH-LHD ist eine erhöhte Expression von Endothelin-β1 mit einem Fortschreiten der Krankheit verbunden und stellt daher ein potenzielles therapeutisches Ziel dar (
89
). ERA sind bei PH-LHD jedoch nur begrenzt wirksam und mit einem hohen Risiko für Nebenwirkungen verbunden (
90
). Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Anämie und Ödeme, die normalerweise dosisabhängig sind (
91
). Bosentan kann Leberfunktionsstörungen verursachen, die sich jedoch normalerweise nach einer Verringerung der Dosis oder einem Absetzen des Medikaments beheben lassen (
92
). Neue ERAs wie Masitentan haben die Häufigkeit dieser Nebenwirkungen durch Optimierung ihrer pharmakokinetischen Eigenschaften verringert (
93
). In der Studie mit Patienten mit fortgeschrittener HFrEF konnte Bosentan den NYHA-Funktionsgrad, den systolischen Pulmonalarteriendruck (PAP), die Geschwindigkeit der Trikuspidalklappeninsuffizienz usw. nicht verbessern, was zu Flüssigkeitsretention und peripheren Ödemen führte und die Hospitalisierungsrate wegen Herzinsuffizienz erhöhte (
94
). Laut der MELODY-1-Studie verbesserte Macitentan PVR, PAWP oder mRAP nach 12-wöchiger Behandlung bei Cpc-PH-Patienten nicht signifikant; der Unterschied war jedoch nicht statistisch signifikant (
95
). Obwohl die Daluxostan-Therapie PAWP, mPAP, PVR oder RAP nicht signifikant senkte, verbesserte sie in der HEAT-Studie den Herzindex bei HF-Patienten nach 3 Wochen. In der Hochdosisgruppe traten Nebenwirkungen häufiger auf. Die EARTH-Studie zeigte auch, dass eine 24-wöchige Behandlung mit Darusentan selbst bei Personen mit echokardiografischem Nachweis von PH den klinischen Zustand oder die kardiale Umgestaltung bei Patienten mit chronischer HF nicht verbesserte. In klinischen Studien zu PH-LHD zeigte ERA generell eine hohe Inzidenz von Nebenwirkungen; mehrere Studien wurden aus Sicherheitsgründen vorzeitig abgebrochen. Daher wird ERA derzeit nicht zur Behandlung von PH-LHD/Cpc-PH empfohlen.Phosphodiesterase (PDE-5-Hemmer)PDE5-Hemmer können die PH und die rechtsventrikuläre Funktion verbessern, indem sie die PDE5-Enzyme hemmen und so zu einer Vasodilatation führen. Ein Cochrane-Review deutet darauf hin, dass die Behandlung mit PDE5-Hemmern die durchschnittliche 6-Minuten-Gehstrecke von Patienten um 48 Meter verlängern kann, während gleichzeitig ihr Funktionsniveau verbessert und das Risiko eines PAH-bedingten Krankenhausaufenthalts gesenkt wird. Die AMBITION-Studie hat gezeigt, dass die anfängliche Kombination von Ambrisentan (ERA) und Tadalafil (PDE5-Hemmer) das Risiko eines klinischen Versagens signifikant senkte. Bei HFrEF-Patienten haben mehrere frühe kleine Studien gezeigt, dass Sildenafil den systolischen pulmonalarteriellen Druck (PASP), mPAP, PVR und 6MWT signifikant senkt und die rechtsventrikuläre Funktion verbessert (
96
,
97
). In einer großen randomisierten kontrollierten Studie ( n = 216) erhöhte Sildenafil im Vergleich zu einer Placebobehandlung weder die körperliche Belastbarkeit, die linksventrikuläre Masse noch kombinierte klinische Endpunkte (Tod, Krankenhausaufenthalt aufgrund von Herz-/Nierenerkrankungen, verstärkte Symptome einer Herzinsuffizienz usw.) . In der Sildenafilgruppe kam es zu einem leichten Anstieg vaskulärer Nebenwirkungen wie Kopfschmerzen, Hitzegefühl und Hypotonie, dieser war jedoch nicht statistisch signifikant (
98
,
99
). Die Wirksamkeit von PDE5-Hemmern bei HFpEF-Patienten ist noch nicht klar, insbesondere bei Patienten ohne rechtsventrikuläre Dysfunktion. Eine rechtsventrikuläre Dysfunktion kann ein wichtiger Prädiktor für den Nutzen einer PDE5-Hemmertherapie sein (
98
). HFpEF-Patienten mit gleichzeitiger Rechtsherzinsuffizienz können von einer PDE5-Hemmertherapie profitieren, da sie beispielsweise den Druck im rechten Vorhof senkt und die Rechtsherzfunktion verbessert. HFpEF-Patienten ohne rechtsventrikuläre Dysfunktion zeigten keinen signifikanten Nutzen (
100
). Mehrere Metaanalysen haben die Wirksamkeit von PDE5-Hemmern (einschließlich Sildenafil) bei PH-LHD zusammengefasst. Bei HFrEF-Patienten verbesserten PDE5-Hemmer den mPAP PVR, die LVEF, die körperliche Leistungsfähigkeit und die Lebensqualität signifikant (
101
,
102
). PDE5-Hemmer wie Sildenafil zeigten bei HFrEF-Patienten bestimmte klinische Vorteile, insbesondere eine Verbesserung der Hämodynamik und der körperlichen Leistungsfähigkeit (
103
). In der SOVIAC-Studie führte Sildenafil bei Patienten mit anhaltender PH nach einer Klappenoperation sogar zu schlechteren klinischen Ergebnissen wie Tod und Krankenhausaufenthalt wegen Herzinsuffizienz (
104
). Die TRITON-Studie untersuchte die Wirksamkeit einer Dreifachkombinationstherapie (Masitentan, Tadalafil und Selexipag) gegenüber einer Zweifachkombinationstherapie (Masitentan und Tadalafil) bei neu diagnostizierten, unbehandelten PH-Patienten, und die Ergebnisse zeigten keinen signifikanten Unterschied beim primären Endpunkt PVR zwischen den beiden. Eine Kohortenanalyse des spanischen PH-Registers untersuchte die prädiktiven Faktoren des Ansprechens auf die Behandlung mit PDE5-Hemmern. Die Ergebnisse zeigten, dass männliches Geschlecht, die Diagnose einer portalen pulmonalen Hypertonie (PoPH) oder HIV-PAH unabhängige Prädiktoren für ein günstiges Ansprechen auf PDE5-Hemmer waren (
105
). Und eine Kohlenmonoxid-Dispersibilität (DLco) ≤ 40 % des vorhergesagten Werts ist mit Nebenwirkungen verbunden. Bei Patienten, die PDE5-Hemmer erhalten haben, aber nicht die gewünschte klinische Wirksamkeit erreicht haben, kann eine Umstellung auf Stimulanzien der löslichen Guanylatcyclase (sGC) in Betracht gezogen werden.Lösliche Guanylcyclase-StimulatorenStimulatoren der löslichen Guanylcyclase fördern die Vasodilatation, indem sie die Produktion von zyklischem Guanosinmonophosphat (cGMP) steigern und so den Mechanismus des PDE5-Hemmers (der durch Verringerung des cGMP-Abbaus wirkt) ergänzen. Sie haben in den letzten Jahren im Bereich der Herzinsuffizienz (HF) und pulmonalen Hypertonie (PH) große Aufmerksamkeit erfahren. In der SOCRATES-REDUCED-Studie wurde Vericiguat von Patienten mit HFrEF gut vertragen, reduzierte jedoch die NT-proBNP-Werte nicht signifikant und verbesserte auch die echokardiographischen Parameter nicht (
106
–
109
). In der SOCRATES-PRESERVED-Studie reduzierte Vericiguat die NT-proBNP-Werte nicht signifikant, war jedoch gut verträglich und mit einer verbesserten Lebensqualität verbunden (
110
). In der VICTORIA-Studie reduzierte Vericiguat den kombinierten Endpunkt aus kardiovaskulärem Tod oder Krankenhauseinweisung wegen Herzinsuffizienz bei Patienten mit sich symptomatischer HFrEF signifikant (
111
). In der LEPHT-Studie wurde Riociguat von Patienten mit HFrEF gut vertragen und verbesserte in der Hochdosisgruppe den Herzindex, den Schlagvolumenindex und den PVR signifikant (
112
). In der DILATE-1-Studie verbesserte Riociguat das Schlagvolumen und den systolischen Blutdruck, hatte jedoch keine signifikante Wirkung auf mPAP und PVR (
113
). In der LEPHT-Studie wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit HFrEF und pulmonaler Hypertonie (PH) untersucht (
114
). Die Studie zeigte, dass die Patienten nach 16-wöchiger Behandlung mit Riociguat (2 mg, dreimal täglich) im Vergleich zur Placebogruppe einen Anstieg des Herzindex und eine signifikante Abnahme des pulmonal-vaskulären Widerstands (PVR) und des systemischen Gefäßwiderstands (SVR) aufwiesen. Die DILATE-1-Studie wurde an Patienten mit HFpEF und PH (LVEF > 50 %, mPAP ≥ 25 mmHg, PAWP > 15 mmHg) durchgeführt. Die Ergebnisse zeigten, dass sich in der Gruppe, die 2 mg Levociguat erhielt, nach 6-stündiger Behandlung das Schlagvolumen und die rechtsventrikuläre enddiastolische Fläche verbesserten, obwohl es zu keiner signifikanten Abnahme des mittleren Pulmonalarteriendrucks (mPAP) kam (
115
). In der Studie COMPERA 2.0 wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit pulmonaler Hypertonie und kardiometabolischen Komorbiditäten untersucht. In der Studie PASSION sollen die Auswirkungen von Riociguat auf die Belastungstoleranz, die Herzfunktionsindizes und die funktionelle WHO-Klassifikation bei Patienten mit Herzinsuffizienz und pulmonaler Hypertonie untersucht werden. In einigen Studien hat Riociguat bei Patienten mit HF-bedingter pulmonaler Hypertonie potenzielle Vorteile gezeigt, wie z. B. verbesserte PVR, SVR und 6MWD, was darauf hindeutet, dass es sich positiv auf die Belastungstoleranz auswirken könnte (
116
). In einer Studie mit 61 Patienten mit pulmonaler Hypertonie (PH), die unzureichend auf PDE-5-Hemmer reagierten, führte die Umstellung auf Riociguat zu einer Erhöhung der 6 MWD nach 24 Wochen, einer Verbesserung des WHO-Grades und einer Senkung der NT-proBNP-Werte (
117
,
118
). Bei Patienten mit Herzinsuffizienz hat Vericiguat bei Patienten mit HFrEF einen signifikanten klinischen Nutzen gezeigt, insbesondere bei der Verringerung des Risikos kardiovaskulärer Ereignisse, hat aber bei Patienten mit HFpEF nur eine begrenzte Wirkung (
119
–
121
). Höhere Dosen von Riociguat verbessern den PVR und den systemischen Gefäßwiderstand bei Patienten mit Cpc-PH, haben aber eine begrenzte Wirksamkeit bei Patienten mit Ipc-PH (
122
,
123
). Die Studie, die Daten aus PATENT-1, PATENT-2, PATENT PLUS und REPLACE umfasste, zeigte, dass Häufigkeit und Schwere der Nebenwirkungen in der Riociguat-Gruppe mit denen in der Placebogruppe vergleichbar waren (
124
).PGI2-MedikamenteProstacyclin wird hauptsächlich von Endothelzellen produziert und hat gefäßerweiternde, antithrombotische und antiproliferative Wirkungen, die die Hämodynamik und die Herzfunktion verbessern können. Zu den Prostacyclin-Medikamenten gehören Prostacyclin-Analoga (wie Epoprostenol, Treprostinil, Iloprost, Beraprost) und Prostacyclin-IP-Rezeptoragonisten (wie Selexipag). Eprostol wirkt, indem es den PVR senkt und die Funktion des rechten Ventrikels verbessert, aber die langfristige Einnahme kann schädliche neurohormonale Systeme (wie das Renin-Angiotensin-System) aktivieren und so eine Herzinsuffizienz verschlimmern. Obwohl Epprostol in der FIRST-Studie den Herzindex, den pulmonalen Verschlussdruck (PAWP) und den systemischen Gefäßwiderstand (SVR) signifikant verbesserte, wurde die Studie aufgrund der verringerten Patientenüberlebensrate vorzeitig abgebrochen. Treprolinil ist ein neuartiges Prostacyclin-Analogon, das den Stoffwechsel und die Herzfunktion verbessern kann, indem es den AMPK-Signalweg in der Skelettmuskulatur und dem rechten Ventrikel aktiviert. In Tiermodellen verbesserte Trepronnier das metabolische Syndrom und senkte den pulmonalarteriellen Druck und zeigte damit das Potenzial, die Entwicklung einer Herzinsuffizienz (HFpEF) mit Ejakulationsfraktionen zu verhindern. Anders als eine Herzinsuffizienz (HFrEF), die eine reduzierte Auswurffraktion aufweist, wird HFpEF häufig von einem metabolischen Syndrom und Diabetes begleitet, und die günstigen Wirkungen von Prostacyclin-Analoga auf den Stoffwechsel und die Lungenblutgefäße machen es möglich, dass es für die Behandlung von PH-HFpEF besser geeignet ist. Bei Patienten mit Cpc-PH verbessert eine gezielte Therapie der pulmonalen Hypertonie (wie Phosphodiesterase-5-Hemmer und Endothelin-Rezeptor-Antagonisten) die Symptome nicht, sondern erhöht stattdessen die Morbiditäts- und Mortalitätsrate. Eine Metaanalyse hat gezeigt, dass eine auf PH ausgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko unerwünschter Ereignisse jedoch höher ist. Eine Metaanalyse hat gezeigt, dass eine pH-zielgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko für Nebenwirkungen jedoch höher ist. Die Wirksamkeit bei menschlichen pH-HFPEF-Patienten muss jedoch noch durch weitere klinische Studien bestätigt werden. Zudem könnte die Behandlungsstrategie der Skelettmuskulatur und des rechtsventrikulären AMPK-Signalwegs eine mögliche Richtung für die Prävention von PH-LHD sein (
125
–
131
).Ralinepag, ein neuartiger Prostazyklin-Rezeptor (IP-Rezeptor)-Agonist, zeigt signifikante gefäßerweiternde Wirkungen, die PVR wirksam reduzieren und gleichzeitig die Proliferation glatter Gefäßmuskelzellen sowie die Thrombozytenaggregation hemmen können. Das Medikament hat eine Halbwertszeit von bis zu 24 Stunden, was ihm einen potenziellen Vorteil bei der Behandlung von PH verschafft. In klinischen Studien der Phase II hat Ralinepag eine gute Wirksamkeit gezeigt. Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Übelkeit und Durchfall. Derzeit wird die klinische Studie der Phase III zu Ralinepag vorangetrieben, und die vorläufigen Langzeitdaten der ADVANCE EXTENSION-Studie wurden auf entsprechenden wissenschaftlichen Konferenzen veröffentlicht (
132
). Derzeit ist die breite Anwendung von Prostazyklin-Medikamenten in der klinischen Praxis aufgrund ihrer geringen Stabilität und vieler Nebenwirkungen begrenzt. Daher ist die Entwicklung neuartiger Prostazyklin-Medikamente mit höherer Rezeptorselektivität, weniger Nebenwirkungen, höherer Stabilität, einfacherer Konservierung und höherer Akzeptanz bei den Patienten zur Forschungsrichtung geworden (
133
).SotatercepSotatercept ist ein Fusionsprotein des Aktivinrezeptors Typ IIA (ActRIIA). Ergebnisse der PULSAR-Studie zeigen, dass Sotatercept Placebo bei der Senkung des PVR und der Verbesserung der 6MWD sowie bei sekundären Endpunkten wie NT-proBNP-Spiegel, funktioneller WHO-Klassifikation und klinischer Verschlechterung deutlich übertrifft. Ergebnisse der STELLAR-Studie zeigen, dass Patienten in der Sotatercept-Gruppe nach 24-wöchiger Behandlung eine Steigerung der 6MWD um 40,8 Meter verzeichneten, verglichen mit einer Steigerung von lediglich 1,0 Meter in der Placebogruppe. Andere sekundäre Endpunkte, darunter Senkungen der NT-proBNP-Spiegel, Verbesserungen der funktionellen WHO-Klassifikation und Verringerung des PVR, waren in der Sotatercept-Gruppe ebenfalls signifikant besser. Bei einer medianen Nachbeobachtungszeit von 32,7 Wochen reduzierte Sotatercept das Risiko einer klinischen Verschlechterung oder des Todes um 84 %. Darüber hinaus verbesserte Sotatercept die Rechtsherzfunktion durch Senkung des mPAP und Verringerung der Arbeitsbelastung der rechten Herzseite. In Bezug auf die Sicherheit umfassten die Nebenwirkungen in der Sotatercept-Gruppe hauptsächlich Kapillarerweiterung, Nasenbluten, Zahnfleischbluten und Thrombozytopenie, die meisten dieser Ereignisse waren jedoch mild. In der Post-hoc- Analyse der STELLAR-Studie wurde festgestellt, dass Sotatercept die Größe der rechten Herzseite reduzierte und die rechtsventrikuläre Funktion sowie die hämodynamischen Parameter nach 24-wöchiger Behandlung verbesserte. Laufende Studien wie SOTERIA, HYPERION, ZENITH, CADENCE, MK-7962-020 und MOONBEAM werden die langfristige Sicherheit und Wirksamkeit von Sotatercept in verschiedenen Patientenpopulationen weiter erforschen und umfassendere klinische Belege für die künftige Behandlung der pulmonalen arteriellen Hypertonie liefern. Im Vergleich zu herkömmlichen niedermolekularen Verbindungen hat Sotatercept eine höhere Wirksamkeit und weniger Nebenwirkungen. Durch die Wiederherstellung des Gleichgewichts des BMPR2-Signalwegs und die Hemmung des Smad2/3-Signalwegs reduziert Sotatercept die Proliferation glatter Lungengefäßmuskelzellen und die Kollagenablagerung und kehrt so den Lungengefäßumbau um. Dieses Ziel ist mit den meisten derzeit verfügbaren zielgerichteten Medikamenten nicht erreichbar. Darüber hinaus sind das Dosierungsschema von Sotatercept von einmal alle drei Wochen und die subkutane Injektionsmethode bequemer als bei herkömmlichen Medikamenten (
Tabelle 4
). Mit seinem einzigartigen Wirkmechanismus und den erheblichen klinischen Vorteilen bietet Sotatercept eine neue Behandlungsoption für Cpc-PH-Patienten und hat das Potenzial, die derzeitige Behandlungslandschaft zu verändern (
134
–
138
). Tabelle 4
Tabelle 3. PGI2-Medikamente. Endothelin-Rezeptor-AntagonistenBisher wurden vier Arten von ERAs klinisch getestet, nämlich Bosentan, Anlisentan, Masitentan und Sitaxentan. Unter diesen wurde Sitaxentan aufgrund tödlicher Leberschäden weltweit vom Markt genommen (
75
). Endothelin-1 (ET-1) ist an der Gefäßverengung und Zellproliferation beteiligt, indem es Endothelinrezeptoren (ET-A und ET- aktiviert (
86
). Cochrane-Studien haben gezeigt, dass die Verwendung von Endothelinrezeptor-Antagonisten über 3 bis 6 Monate die körperliche Leistungsfähigkeit, die Symptome und die kardiorespiratorischen hämodynamischen Indikatoren der Patienten signifikant verbessert (
87
). Derzeit ist jedoch unklar, ob diese Medikamente die Sterblichkeitsrate der Patienten signifikant senken können (
88
). Bei PH-LHD ist eine erhöhte Expression von Endothelin-β1 mit einem Fortschreiten der Krankheit verbunden und stellt daher ein potenzielles therapeutisches Ziel dar (
89
). ERA sind bei PH-LHD jedoch nur begrenzt wirksam und mit einem hohen Risiko für Nebenwirkungen verbunden (
90
). Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Anämie und Ödeme, die normalerweise dosisabhängig sind (
91
). Bosentan kann Leberfunktionsstörungen verursachen, die sich jedoch normalerweise nach einer Verringerung der Dosis oder einem Absetzen des Medikaments beheben lassen (
92
). Neue ERAs wie Masitentan haben die Häufigkeit dieser Nebenwirkungen durch Optimierung ihrer pharmakokinetischen Eigenschaften verringert (
93
). In der Studie mit Patienten mit fortgeschrittener HFrEF konnte Bosentan den NYHA-Funktionsgrad, den systolischen Pulmonalarteriendruck (PAP), die Geschwindigkeit der Trikuspidalklappeninsuffizienz usw. nicht verbessern, was zu Flüssigkeitsretention und peripheren Ödemen führte und die Hospitalisierungsrate wegen Herzinsuffizienz erhöhte (
94
). Laut der MELODY-1-Studie verbesserte Macitentan PVR, PAWP oder mRAP nach 12-wöchiger Behandlung bei Cpc-PH-Patienten nicht signifikant; der Unterschied war jedoch nicht statistisch signifikant (
95
). Obwohl die Daluxostan-Therapie PAWP, mPAP, PVR oder RAP nicht signifikant senkte, verbesserte sie in der HEAT-Studie den Herzindex bei HF-Patienten nach 3 Wochen. In der Hochdosisgruppe traten Nebenwirkungen häufiger auf. Die EARTH-Studie zeigte auch, dass eine 24-wöchige Behandlung mit Darusentan selbst bei Personen mit echokardiografischem Nachweis von PH den klinischen Zustand oder die kardiale Umgestaltung bei Patienten mit chronischer HF nicht verbesserte. In klinischen Studien zu PH-LHD zeigte ERA generell eine hohe Inzidenz von Nebenwirkungen; mehrere Studien wurden aus Sicherheitsgründen vorzeitig abgebrochen. Daher wird ERA derzeit nicht zur Behandlung von PH-LHD/Cpc-PH empfohlen.Phosphodiesterase (PDE-5-Hemmer)PDE5-Hemmer können die PH und die rechtsventrikuläre Funktion verbessern, indem sie die PDE5-Enzyme hemmen und so zu einer Vasodilatation führen. Ein Cochrane-Review deutet darauf hin, dass die Behandlung mit PDE5-Hemmern die durchschnittliche 6-Minuten-Gehstrecke von Patienten um 48 Meter verlängern kann, während gleichzeitig ihr Funktionsniveau verbessert und das Risiko eines PAH-bedingten Krankenhausaufenthalts gesenkt wird. Die AMBITION-Studie hat gezeigt, dass die anfängliche Kombination von Ambrisentan (ERA) und Tadalafil (PDE5-Hemmer) das Risiko eines klinischen Versagens signifikant senkte. Bei HFrEF-Patienten haben mehrere frühe kleine Studien gezeigt, dass Sildenafil den systolischen pulmonalarteriellen Druck (PASP), mPAP, PVR und 6MWT signifikant senkt und die rechtsventrikuläre Funktion verbessert (
96
,
97
). In einer großen randomisierten kontrollierten Studie ( n = 216) erhöhte Sildenafil im Vergleich zu einer Placebobehandlung weder die körperliche Belastbarkeit, die linksventrikuläre Masse noch kombinierte klinische Endpunkte (Tod, Krankenhausaufenthalt aufgrund von Herz-/Nierenerkrankungen, verstärkte Symptome einer Herzinsuffizienz usw.) . In der Sildenafilgruppe kam es zu einem leichten Anstieg vaskulärer Nebenwirkungen wie Kopfschmerzen, Hitzegefühl und Hypotonie, dieser war jedoch nicht statistisch signifikant (
98
,
99
). Die Wirksamkeit von PDE5-Hemmern bei HFpEF-Patienten ist noch nicht klar, insbesondere bei Patienten ohne rechtsventrikuläre Dysfunktion. Eine rechtsventrikuläre Dysfunktion kann ein wichtiger Prädiktor für den Nutzen einer PDE5-Hemmertherapie sein (
98
). HFpEF-Patienten mit gleichzeitiger Rechtsherzinsuffizienz können von einer PDE5-Hemmertherapie profitieren, da sie beispielsweise den Druck im rechten Vorhof senkt und die Rechtsherzfunktion verbessert. HFpEF-Patienten ohne rechtsventrikuläre Dysfunktion zeigten keinen signifikanten Nutzen (
100
). Mehrere Metaanalysen haben die Wirksamkeit von PDE5-Hemmern (einschließlich Sildenafil) bei PH-LHD zusammengefasst. Bei HFrEF-Patienten verbesserten PDE5-Hemmer den mPAP PVR, die LVEF, die körperliche Leistungsfähigkeit und die Lebensqualität signifikant (
101
,
102
). PDE5-Hemmer wie Sildenafil zeigten bei HFrEF-Patienten bestimmte klinische Vorteile, insbesondere eine Verbesserung der Hämodynamik und der körperlichen Leistungsfähigkeit (
103
). In der SOVIAC-Studie führte Sildenafil bei Patienten mit anhaltender PH nach einer Klappenoperation sogar zu schlechteren klinischen Ergebnissen wie Tod und Krankenhausaufenthalt wegen Herzinsuffizienz (
104
). Die TRITON-Studie untersuchte die Wirksamkeit einer Dreifachkombinationstherapie (Masitentan, Tadalafil und Selexipag) gegenüber einer Zweifachkombinationstherapie (Masitentan und Tadalafil) bei neu diagnostizierten, unbehandelten PH-Patienten, und die Ergebnisse zeigten keinen signifikanten Unterschied beim primären Endpunkt PVR zwischen den beiden. Eine Kohortenanalyse des spanischen PH-Registers untersuchte die prädiktiven Faktoren des Ansprechens auf die Behandlung mit PDE5-Hemmern. Die Ergebnisse zeigten, dass männliches Geschlecht, die Diagnose einer portalen pulmonalen Hypertonie (PoPH) oder HIV-PAH unabhängige Prädiktoren für ein günstiges Ansprechen auf PDE5-Hemmer waren (
105
). Und eine Kohlenmonoxid-Dispersibilität (DLco) ≤ 40 % des vorhergesagten Werts ist mit Nebenwirkungen verbunden. Bei Patienten, die PDE5-Hemmer erhalten haben, aber nicht die gewünschte klinische Wirksamkeit erreicht haben, kann eine Umstellung auf Stimulanzien der löslichen Guanylatcyclase (sGC) in Betracht gezogen werden.Lösliche Guanylcyclase-StimulatorenStimulatoren der löslichen Guanylcyclase fördern die Vasodilatation, indem sie die Produktion von zyklischem Guanosinmonophosphat (cGMP) steigern und so den Mechanismus des PDE5-Hemmers (der durch Verringerung des cGMP-Abbaus wirkt) ergänzen. Sie haben in den letzten Jahren im Bereich der Herzinsuffizienz (HF) und pulmonalen Hypertonie (PH) große Aufmerksamkeit erfahren. In der SOCRATES-REDUCED-Studie wurde Vericiguat von Patienten mit HFrEF gut vertragen, reduzierte jedoch die NT-proBNP-Werte nicht signifikant und verbesserte auch die echokardiographischen Parameter nicht (
106
–
109
). In der SOCRATES-PRESERVED-Studie reduzierte Vericiguat die NT-proBNP-Werte nicht signifikant, war jedoch gut verträglich und mit einer verbesserten Lebensqualität verbunden (
110
). In der VICTORIA-Studie reduzierte Vericiguat den kombinierten Endpunkt aus kardiovaskulärem Tod oder Krankenhauseinweisung wegen Herzinsuffizienz bei Patienten mit sich symptomatischer HFrEF signifikant (
111
). In der LEPHT-Studie wurde Riociguat von Patienten mit HFrEF gut vertragen und verbesserte in der Hochdosisgruppe den Herzindex, den Schlagvolumenindex und den PVR signifikant (
112
). In der DILATE-1-Studie verbesserte Riociguat das Schlagvolumen und den systolischen Blutdruck, hatte jedoch keine signifikante Wirkung auf mPAP und PVR (
113
). In der LEPHT-Studie wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit HFrEF und pulmonaler Hypertonie (PH) untersucht (
114
). Die Studie zeigte, dass die Patienten nach 16-wöchiger Behandlung mit Riociguat (2 mg, dreimal täglich) im Vergleich zur Placebogruppe einen Anstieg des Herzindex und eine signifikante Abnahme des pulmonal-vaskulären Widerstands (PVR) und des systemischen Gefäßwiderstands (SVR) aufwiesen. Die DILATE-1-Studie wurde an Patienten mit HFpEF und PH (LVEF > 50 %, mPAP ≥ 25 mmHg, PAWP > 15 mmHg) durchgeführt. Die Ergebnisse zeigten, dass sich in der Gruppe, die 2 mg Levociguat erhielt, nach 6-stündiger Behandlung das Schlagvolumen und die rechtsventrikuläre enddiastolische Fläche verbesserten, obwohl es zu keiner signifikanten Abnahme des mittleren Pulmonalarteriendrucks (mPAP) kam (
115
). In der Studie COMPERA 2.0 wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit pulmonaler Hypertonie und kardiometabolischen Komorbiditäten untersucht. In der Studie PASSION sollen die Auswirkungen von Riociguat auf die Belastungstoleranz, die Herzfunktionsindizes und die funktionelle WHO-Klassifikation bei Patienten mit Herzinsuffizienz und pulmonaler Hypertonie untersucht werden. In einigen Studien hat Riociguat bei Patienten mit HF-bedingter pulmonaler Hypertonie potenzielle Vorteile gezeigt, wie z. B. verbesserte PVR, SVR und 6MWD, was darauf hindeutet, dass es sich positiv auf die Belastungstoleranz auswirken könnte (
116
). In einer Studie mit 61 Patienten mit pulmonaler Hypertonie (PH), die unzureichend auf PDE-5-Hemmer reagierten, führte die Umstellung auf Riociguat zu einer Erhöhung der 6 MWD nach 24 Wochen, einer Verbesserung des WHO-Grades und einer Senkung der NT-proBNP-Werte (
117
,
118
). Bei Patienten mit Herzinsuffizienz hat Vericiguat bei Patienten mit HFrEF einen signifikanten klinischen Nutzen gezeigt, insbesondere bei der Verringerung des Risikos kardiovaskulärer Ereignisse, hat aber bei Patienten mit HFpEF nur eine begrenzte Wirkung (
119
–
121
). Höhere Dosen von Riociguat verbessern den PVR und den systemischen Gefäßwiderstand bei Patienten mit Cpc-PH, haben aber eine begrenzte Wirksamkeit bei Patienten mit Ipc-PH (
122
,
123
). Die Studie, die Daten aus PATENT-1, PATENT-2, PATENT PLUS und REPLACE umfasste, zeigte, dass Häufigkeit und Schwere der Nebenwirkungen in der Riociguat-Gruppe mit denen in der Placebogruppe vergleichbar waren (
124
).PGI2-MedikamenteProstacyclin wird hauptsächlich von Endothelzellen produziert und hat gefäßerweiternde, antithrombotische und antiproliferative Wirkungen, die die Hämodynamik und die Herzfunktion verbessern können. Zu den Prostacyclin-Medikamenten gehören Prostacyclin-Analoga (wie Epoprostenol, Treprostinil, Iloprost, Beraprost) und Prostacyclin-IP-Rezeptoragonisten (wie Selexipag). Eprostol wirkt, indem es den PVR senkt und die Funktion des rechten Ventrikels verbessert, aber die langfristige Einnahme kann schädliche neurohormonale Systeme (wie das Renin-Angiotensin-System) aktivieren und so eine Herzinsuffizienz verschlimmern. Obwohl Epprostol in der FIRST-Studie den Herzindex, den pulmonalen Verschlussdruck (PAWP) und den systemischen Gefäßwiderstand (SVR) signifikant verbesserte, wurde die Studie aufgrund der verringerten Patientenüberlebensrate vorzeitig abgebrochen. Treprolinil ist ein neuartiges Prostacyclin-Analogon, das den Stoffwechsel und die Herzfunktion verbessern kann, indem es den AMPK-Signalweg in der Skelettmuskulatur und dem rechten Ventrikel aktiviert. In Tiermodellen verbesserte Trepronnier das metabolische Syndrom und senkte den pulmonalarteriellen Druck und zeigte damit das Potenzial, die Entwicklung einer Herzinsuffizienz (HFpEF) mit Ejakulationsfraktionen zu verhindern. Anders als eine Herzinsuffizienz (HFrEF), die eine reduzierte Auswurffraktion aufweist, wird HFpEF häufig von einem metabolischen Syndrom und Diabetes begleitet, und die günstigen Wirkungen von Prostacyclin-Analoga auf den Stoffwechsel und die Lungenblutgefäße machen es möglich, dass es für die Behandlung von PH-HFpEF besser geeignet ist. Bei Patienten mit Cpc-PH verbessert eine gezielte Therapie der pulmonalen Hypertonie (wie Phosphodiesterase-5-Hemmer und Endothelin-Rezeptor-Antagonisten) die Symptome nicht, sondern erhöht stattdessen die Morbiditäts- und Mortalitätsrate. Eine Metaanalyse hat gezeigt, dass eine auf PH ausgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko unerwünschter Ereignisse jedoch höher ist. Eine Metaanalyse hat gezeigt, dass eine pH-zielgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko für Nebenwirkungen jedoch höher ist. Die Wirksamkeit bei menschlichen pH-HFPEF-Patienten muss jedoch noch durch weitere klinische Studien bestätigt werden. Zudem könnte die Behandlungsstrategie der Skelettmuskulatur und des rechtsventrikulären AMPK-Signalwegs eine mögliche Richtung für die Prävention von PH-LHD sein (
125
–
131
).Ralinepag, ein neuartiger Prostazyklin-Rezeptor (IP-Rezeptor)-Agonist, zeigt signifikante gefäßerweiternde Wirkungen, die PVR wirksam reduzieren und gleichzeitig die Proliferation glatter Gefäßmuskelzellen sowie die Thrombozytenaggregation hemmen können. Das Medikament hat eine Halbwertszeit von bis zu 24 Stunden, was ihm einen potenziellen Vorteil bei der Behandlung von PH verschafft. In klinischen Studien der Phase II hat Ralinepag eine gute Wirksamkeit gezeigt. Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Übelkeit und Durchfall. Derzeit wird die klinische Studie der Phase III zu Ralinepag vorangetrieben, und die vorläufigen Langzeitdaten der ADVANCE EXTENSION-Studie wurden auf entsprechenden wissenschaftlichen Konferenzen veröffentlicht (
132
). Derzeit ist die breite Anwendung von Prostazyklin-Medikamenten in der klinischen Praxis aufgrund ihrer geringen Stabilität und vieler Nebenwirkungen begrenzt. Daher ist die Entwicklung neuartiger Prostazyklin-Medikamente mit höherer Rezeptorselektivität, weniger Nebenwirkungen, höherer Stabilität, einfacherer Konservierung und höherer Akzeptanz bei den Patienten zur Forschungsrichtung geworden (
133
).SotatercepSotatercept ist ein Fusionsprotein des Aktivinrezeptors Typ IIA (ActRIIA). Ergebnisse der PULSAR-Studie zeigen, dass Sotatercept Placebo bei der Senkung des PVR und der Verbesserung der 6MWD sowie bei sekundären Endpunkten wie NT-proBNP-Spiegel, funktioneller WHO-Klassifikation und klinischer Verschlechterung deutlich übertrifft. Ergebnisse der STELLAR-Studie zeigen, dass Patienten in der Sotatercept-Gruppe nach 24-wöchiger Behandlung eine Steigerung der 6MWD um 40,8 Meter verzeichneten, verglichen mit einer Steigerung von lediglich 1,0 Meter in der Placebogruppe. Andere sekundäre Endpunkte, darunter Senkungen der NT-proBNP-Spiegel, Verbesserungen der funktionellen WHO-Klassifikation und Verringerung des PVR, waren in der Sotatercept-Gruppe ebenfalls signifikant besser. Bei einer medianen Nachbeobachtungszeit von 32,7 Wochen reduzierte Sotatercept das Risiko einer klinischen Verschlechterung oder des Todes um 84 %. Darüber hinaus verbesserte Sotatercept die Rechtsherzfunktion durch Senkung des mPAP und Verringerung der Arbeitsbelastung der rechten Herzseite. In Bezug auf die Sicherheit umfassten die Nebenwirkungen in der Sotatercept-Gruppe hauptsächlich Kapillarerweiterung, Nasenbluten, Zahnfleischbluten und Thrombozytopenie, die meisten dieser Ereignisse waren jedoch mild. In der Post-hoc- Analyse der STELLAR-Studie wurde festgestellt, dass Sotatercept die Größe der rechten Herzseite reduzierte und die rechtsventrikuläre Funktion sowie die hämodynamischen Parameter nach 24-wöchiger Behandlung verbesserte. Laufende Studien wie SOTERIA, HYPERION, ZENITH, CADENCE, MK-7962-020 und MOONBEAM werden die langfristige Sicherheit und Wirksamkeit von Sotatercept in verschiedenen Patientenpopulationen weiter erforschen und umfassendere klinische Belege für die künftige Behandlung der pulmonalen arteriellen Hypertonie liefern. Im Vergleich zu herkömmlichen niedermolekularen Verbindungen hat Sotatercept eine höhere Wirksamkeit und weniger Nebenwirkungen. Durch die Wiederherstellung des Gleichgewichts des BMPR2-Signalwegs und die Hemmung des Smad2/3-Signalwegs reduziert Sotatercept die Proliferation glatter Lungengefäßmuskelzellen und die Kollagenablagerung und kehrt so den Lungengefäßumbau um. Dieses Ziel ist mit den meisten derzeit verfügbaren zielgerichteten Medikamenten nicht erreichbar. Darüber hinaus sind das Dosierungsschema von Sotatercept von einmal alle drei Wochen und die subkutane Injektionsmethode bequemer als bei herkömmlichen Medikamenten (
Tabelle 4
). Mit seinem einzigartigen Wirkmechanismus und den erheblichen klinischen Vorteilen bietet Sotatercept eine neue Behandlungsoption für Cpc-PH-Patienten und hat das Potenzial, die derzeitige Behandlungslandschaft zu verändern (
134
–
138
). Tabelle 4
 Tabelle 4. Sotatercept. Laufende klinische StudienViele Patienten mit Herz-Kreislauf-Erkrankungen leiden häufig unter dem Metabolischen Syndrom (MS), das unter anderem mit Fettleibigkeit, Dyslipidämie, Insulinresistenz und Diabetes einhergeht (
Tabelle 5
). MS kann systemische Entzündungsreaktionen auslösen, und die Infiltration von Entzündungsfaktoren sowie ein Ungleichgewicht in der Immunmodulation sind wichtige pathogene Faktoren für den Gefäßumbau. Daher könnte die Immuntherapie eine neue Therapiestrategie für PH-LHD/Cpc-PH werden (
139
–
142
) (
Tabelle 6
). Tabelle 5
Tabelle 4. Sotatercept. Laufende klinische StudienViele Patienten mit Herz-Kreislauf-Erkrankungen leiden häufig unter dem Metabolischen Syndrom (MS), das unter anderem mit Fettleibigkeit, Dyslipidämie, Insulinresistenz und Diabetes einhergeht (
Tabelle 5
). MS kann systemische Entzündungsreaktionen auslösen, und die Infiltration von Entzündungsfaktoren sowie ein Ungleichgewicht in der Immunmodulation sind wichtige pathogene Faktoren für den Gefäßumbau. Daher könnte die Immuntherapie eine neue Therapiestrategie für PH-LHD/Cpc-PH werden (
139
–
142
) (
Tabelle 6
). Tabelle 5
 Tabelle 5. Laufende klinische Studien. Tabelle 6
Tabelle 5. Laufende klinische Studien. Tabelle 6
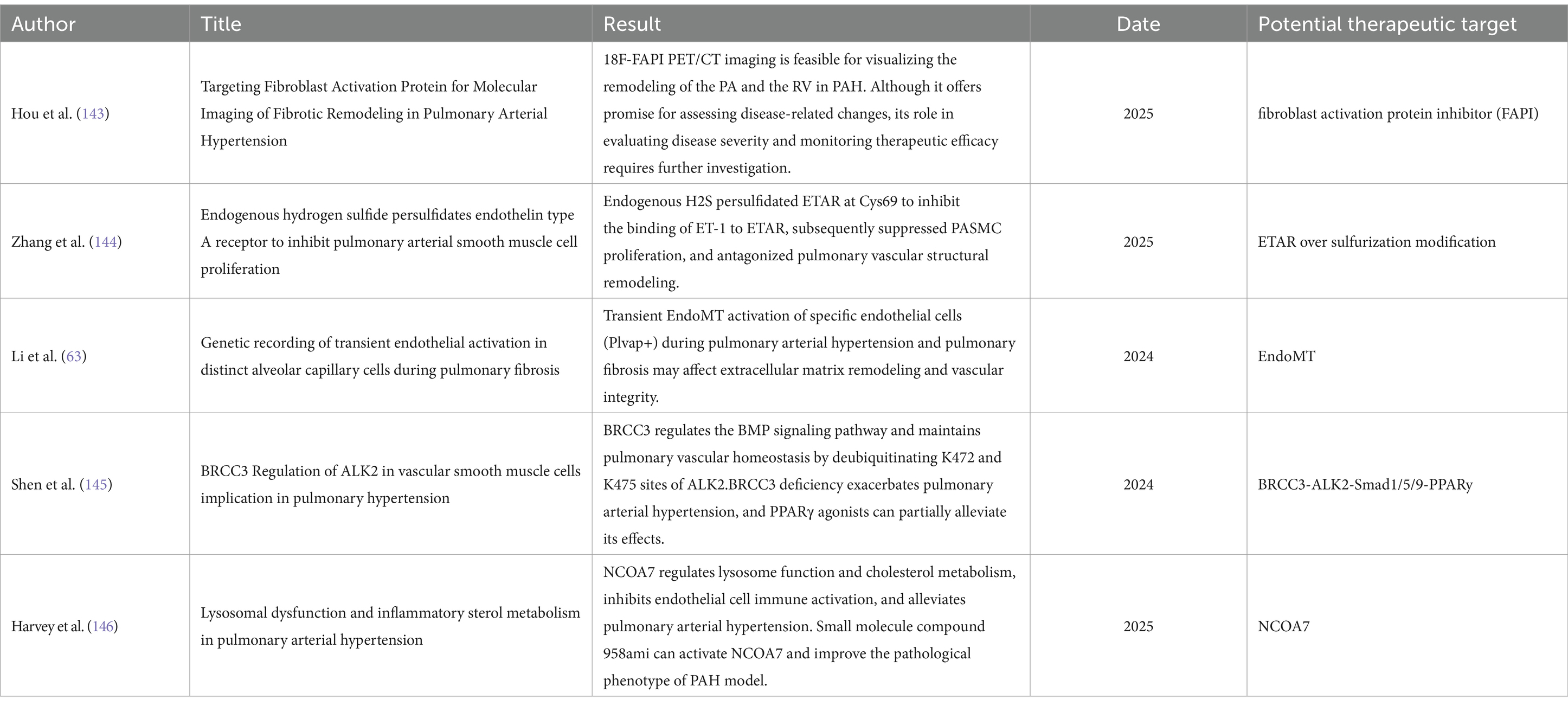 Tabelle 6. Studien zur Aufdeckung potenzieller therapeutischer Ziele. Potenzielle neue therapeutische ZieleDie Stammzelltherapie hat als neuer therapeutischer Ansatz aufgrund ihrer Fähigkeit zur Selbsterneuerung, Proliferation und Differenzierung in eine Vielzahl spezifischer Zellen viel Aufmerksamkeit erregt. Zu den Stammzellen, die häufig zur Behandlung von pulmonaler Hypertonie (PH) eingesetzt werden, gehören endotheliale Progenitorzellen (EPCs) und mesenchymale Stammzellen (MSCs). Unter diesen sind EPCs oligonische Stammzellen, die dazu neigen, sich in Endothelzellen (EC) zu differenzieren, in einem pathologischen Zustand jedoch das Auftreten und die Entwicklung von PH fördern können. Beispielsweise zeigten EPCs in hypoxischen neugeborenen Kälbern eine stärkere Migration und Gangkapazität. Um die Defekte der EPCs zu korrigieren, ist die Zellinfusion ein direkter Ansatz. Mit fortschreitender PH können EPCs jedoch ihre hämatopoetischen Tendenzen reaktivieren, indem sie hämatopoetische Transkriptionsfaktoren abnorm exprimieren, was zu einer übermäßigen Infiltration von Immunzellen führt. Liang et al. blockierten erfolgreich den EHT-Prozess und kehrten den Rückgang der EPC-Werte um, indem sie den Endothel-Hämatopoietischen-Transition-(EHT)-Inhibitor Runx1 in einem monoklonalen Toxin (MCT) mit Einzeldosis einsetzten. Darüber hinaus wurde gezeigt, dass Erythropoietin (EPO) gefäßschützende Wirkungen hat, indem es die Anzahl der zirkulierenden EPCs wiederherstellt und den Gefäßumbau umkehrt, indem es die Expression von Hämoxygenase-1 (HO-1) fördert. Mesenchymale Stammzellen (MSCs) sind pluripotente Stammzellen, die dazu neigen, sich in mesenchymale Zellen wie Osteoblasten, Adipozyten, Chondrozyten und Myozyten zu differenzieren. MSCs zeigen ein erhebliches Potenzial in der Behandlung von PH, insbesondere in Rattenmodellen, die durch chronische Hypoxie und die Gabe eines monoklonalen Toxins (SuHx) induziert wurden. Dort sind MSCs in der Lage, die Kollagenablagerung umzukehren und peripulmonale vaskulär-hämophile Faktoren (vWF) sowie α -Glattmuskelaktin (α-SMA) zu reduzieren. Darüber hinaus lindern MSCs die PH-Symptome, indem sie die Darmflora regulieren. Studien haben gezeigt, dass sowohl SuHx als auch MCT die Homöostase der Darmmikrobiota bei Mäusen zerstören können, während die Behandlung mit MSCs den entzündungshemmenden Bakterienspiegel wiederherstellen und die immunregulierende funktionelle Bakterienpopulation verbessern kann (
147
–
151
).Fortschritte in der interventionellen BehandlungEine neue interventionelle Therapietechnik namens Pulmonalarterien-Denervierung (PADN), die auf Katheterablation beruht, reduziert den Pulmonalarterienwiderstand, verbessert die Rechtsherzfunktion und verzögert die Pulmonalarterienerkrankung durch Hemmung der sympathischen Nervenaktivität durch Ablation nervenintensiver Bereiche in oder nahe der Pulmonalarterie. Fortschritt bei Hochdruck (PH). Sowohl klinische Forschung als auch Tierversuche haben positive Ergebnisse erbracht. Jüngsten Forschungsergebnissen zufolge kann PADN die Downregulation β -adrenerger Rezeptoren und die Überexpression α -adrenerger Rezeptoren in Rattenlungengewebe im präklinischen Modell der pulmonalen Hypertonie-Linksherzerkrankung (PH-LHD) umkehren. PADN senkte in einer Phase-II-Studie mit PH gemischter Ätiologie, einschließlich PH-LHD, signifikant den mittleren pulmonalarteriellen Druck (mPAP) und die 6-Minuten-Gehstrecke. Multizentrische Studien zu Cpc-PH zeigten zudem, dass PADN den systolischen und diastolischen Pulmonalarteriendruck senken, das Herzzeitvolumen und die 6-Minuten-Gehstrecke erhöhen kann, während gleichzeitig die klinische Verschlechterung und die Hospitalisierungsrate signifikant reduziert werden. Ein
Tabelle 6. Studien zur Aufdeckung potenzieller therapeutischer Ziele. Potenzielle neue therapeutische ZieleDie Stammzelltherapie hat als neuer therapeutischer Ansatz aufgrund ihrer Fähigkeit zur Selbsterneuerung, Proliferation und Differenzierung in eine Vielzahl spezifischer Zellen viel Aufmerksamkeit erregt. Zu den Stammzellen, die häufig zur Behandlung von pulmonaler Hypertonie (PH) eingesetzt werden, gehören endotheliale Progenitorzellen (EPCs) und mesenchymale Stammzellen (MSCs). Unter diesen sind EPCs oligonische Stammzellen, die dazu neigen, sich in Endothelzellen (EC) zu differenzieren, in einem pathologischen Zustand jedoch das Auftreten und die Entwicklung von PH fördern können. Beispielsweise zeigten EPCs in hypoxischen neugeborenen Kälbern eine stärkere Migration und Gangkapazität. Um die Defekte der EPCs zu korrigieren, ist die Zellinfusion ein direkter Ansatz. Mit fortschreitender PH können EPCs jedoch ihre hämatopoetischen Tendenzen reaktivieren, indem sie hämatopoetische Transkriptionsfaktoren abnorm exprimieren, was zu einer übermäßigen Infiltration von Immunzellen führt. Liang et al. blockierten erfolgreich den EHT-Prozess und kehrten den Rückgang der EPC-Werte um, indem sie den Endothel-Hämatopoietischen-Transition-(EHT)-Inhibitor Runx1 in einem monoklonalen Toxin (MCT) mit Einzeldosis einsetzten. Darüber hinaus wurde gezeigt, dass Erythropoietin (EPO) gefäßschützende Wirkungen hat, indem es die Anzahl der zirkulierenden EPCs wiederherstellt und den Gefäßumbau umkehrt, indem es die Expression von Hämoxygenase-1 (HO-1) fördert. Mesenchymale Stammzellen (MSCs) sind pluripotente Stammzellen, die dazu neigen, sich in mesenchymale Zellen wie Osteoblasten, Adipozyten, Chondrozyten und Myozyten zu differenzieren. MSCs zeigen ein erhebliches Potenzial in der Behandlung von PH, insbesondere in Rattenmodellen, die durch chronische Hypoxie und die Gabe eines monoklonalen Toxins (SuHx) induziert wurden. Dort sind MSCs in der Lage, die Kollagenablagerung umzukehren und peripulmonale vaskulär-hämophile Faktoren (vWF) sowie α -Glattmuskelaktin (α-SMA) zu reduzieren. Darüber hinaus lindern MSCs die PH-Symptome, indem sie die Darmflora regulieren. Studien haben gezeigt, dass sowohl SuHx als auch MCT die Homöostase der Darmmikrobiota bei Mäusen zerstören können, während die Behandlung mit MSCs den entzündungshemmenden Bakterienspiegel wiederherstellen und die immunregulierende funktionelle Bakterienpopulation verbessern kann (
147
–
151
).Fortschritte in der interventionellen BehandlungEine neue interventionelle Therapietechnik namens Pulmonalarterien-Denervierung (PADN), die auf Katheterablation beruht, reduziert den Pulmonalarterienwiderstand, verbessert die Rechtsherzfunktion und verzögert die Pulmonalarterienerkrankung durch Hemmung der sympathischen Nervenaktivität durch Ablation nervenintensiver Bereiche in oder nahe der Pulmonalarterie. Fortschritt bei Hochdruck (PH). Sowohl klinische Forschung als auch Tierversuche haben positive Ergebnisse erbracht. Jüngsten Forschungsergebnissen zufolge kann PADN die Downregulation β -adrenerger Rezeptoren und die Überexpression α -adrenerger Rezeptoren in Rattenlungengewebe im präklinischen Modell der pulmonalen Hypertonie-Linksherzerkrankung (PH-LHD) umkehren. PADN senkte in einer Phase-II-Studie mit PH gemischter Ätiologie, einschließlich PH-LHD, signifikant den mittleren pulmonalarteriellen Druck (mPAP) und die 6-Minuten-Gehstrecke. Multizentrische Studien zu Cpc-PH zeigten zudem, dass PADN den systolischen und diastolischen Pulmonalarteriendruck senken, das Herzzeitvolumen und die 6-Minuten-Gehstrecke erhöhen kann, während gleichzeitig die klinische Verschlechterung und die Hospitalisierungsrate signifikant reduziert werden. Ein
und seine Rezeptoren CXCR1 und CXCR2 sind bei pulmonaler arterieller Hypertonie hochreguliert, wobei CXCL8 durch Bindung an CXCR1/2 die Rekrutierung und Aktivierung von Entzündungszellen fördert und so die Entzündung und den Umbau der Pulmonalarterie verschlimmert. Die Interaktion zwischen CXCL10 und seinem Rezeptor CXCR3 spielt eine bedeutende Rolle in der Pathogenese der PH, wobei CXCL10 die Chemotaxis von T-Zellen und natürlichen Killerzellen induziert und an der Entzündungsreaktion der Pulmonalarterie beteiligt ist. Die Signalwege von CXCL12 und seinen Rezeptoren CXCR4 und ACKR3 spielen ebenfalls wichtige Rollen bei PH, wobei die CXCL12-CXCR4-Achse nicht nur die Migration von Entzündungszellen reguliert, sondern auch die Proliferation von PASMCs über den PI3K/Akt-Signalweg fördert (
33
–
36
).Genetische Faktoren und molekulare SignalwegmechanismenBMPR-II ist ein Mitglied der TGF- β- Superfamilie. Heterozygote Mutationen finden sich bei 80 % der Patienten mit familiärer PH und 20 % der Patienten mit sporadischer PH, was es zu einem pathogenen Schlüsselgen für primäre und hereditäre PH macht (
37
–
39
). Es beeinflusst Zellproliferation, Differenzierung, Gewebereparatur, Entzündung und Angiogenese über Smad1/5/8 und nicht-Smad-Signalwege. Die Mutationsrate des BMP9-Gens beträgt in ostasiatischen Populationen bis zu 6,7 %. Die Mutationen reduzieren den zirkulierenden BMP9-Spiegel, schwächen die vaskulären Fähigkeiten zur Apoptose und Schädigung und erhöhen das Risiko für idiopathische PH um mehr als das 22-Fache (
40
–
42
). Der PPARγ-p53-Signalweg reguliert Zellproliferation, Apoptose und Entzündungsreaktionen und beeinflusst die pulmonale Gefäßumgestaltung (
43
–
47
). Auch andere Genmutationen (wie SMAD1, SMAD4, SMAD9, CAV1 und KCNK3) wurden in BMPR-II-Mutations-negativen Familien identifiziert (
48
–
50
). Unter Stressbedingungen wird CLIC4 stark exprimiert, was die Internalisierung und den Abbau von BMPR-II fördert, den BMP-Signalweg hemmt und eine phänotypische Verschiebung in Endothelzellen in Richtung antiapoptotischer und proliferativer Zustände induziert (
51
–
57
).Bei der eingehenden Untersuchung der Pathogenese von Cpc-PH deuten immer mehr Hinweise darauf hin, dass Mutationen in mehreren funktionellen Genen (wie BMPRII, ALK1, CAV1, TBX4, KCNK3, SMAD8, EIF2AK4 usw.) eine wichtige Rolle bei der Pathogenese der Krankheit spielen. Derzeit gehen Forscher im Allgemeinen davon aus, dass das Auftreten von PH ähnlich wie bei Tumoren verläuft, mit einem „Sekundärschlag“-Mechanismus, bei dem genetische und Umweltfaktoren gemeinsam das Auftreten und die Entwicklung der Krankheit fördern.Gentherapie und molekulare SignalwegmechanismenExperimente mit p53-Nanopartikeln und AAV-Vektor-vermittelter Freisetzung normaler BMPR2-Gene haben vielversprechende Ergebnisse gezeigt. Lipid-Nanopartikel (LNPs), die CRISPR-Cas9-Systeme freisetzen, können Genmutationen in PASMCs reparieren (
58
–
60
). Canagliflozin lindert PH-Symptome durch Aktivierung von PPARγ und Hemmung seiner S225-Phosphorylierung und zeigt in Tiermodellen gute Schutzeffekte. Die Aufrechterhaltung der pulmonalvaskulären Homöostase durch Deubiquitinierung zur Aktivierung der ALK2-Smad1/5/9-PPARγ-Achse mit Hochregulierung von BRCC3 kann PH-Symptome lindern (
61
–
64
) (
Abbildung 2
). Abbildung 2
aktiviert (
86
). Cochrane-Studien haben gezeigt, dass die Verwendung von Endothelinrezeptor-Antagonisten über 3 bis 6 Monate die körperliche Leistungsfähigkeit, die Symptome und die kardiorespiratorischen hämodynamischen Indikatoren der Patienten signifikant verbessert (
87
). Derzeit ist jedoch unklar, ob diese Medikamente die Sterblichkeitsrate der Patienten signifikant senken können (
88
). Bei PH-LHD ist eine erhöhte Expression von Endothelin-β1 mit einem Fortschreiten der Krankheit verbunden und stellt daher ein potenzielles therapeutisches Ziel dar (
89
). ERA sind bei PH-LHD jedoch nur begrenzt wirksam und mit einem hohen Risiko für Nebenwirkungen verbunden (
90
). Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Anämie und Ödeme, die normalerweise dosisabhängig sind (
91
). Bosentan kann Leberfunktionsstörungen verursachen, die sich jedoch normalerweise nach einer Verringerung der Dosis oder einem Absetzen des Medikaments beheben lassen (
92
). Neue ERAs wie Masitentan haben die Häufigkeit dieser Nebenwirkungen durch Optimierung ihrer pharmakokinetischen Eigenschaften verringert (
93
). In der Studie mit Patienten mit fortgeschrittener HFrEF konnte Bosentan den NYHA-Funktionsgrad, den systolischen Pulmonalarteriendruck (PAP), die Geschwindigkeit der Trikuspidalklappeninsuffizienz usw. nicht verbessern, was zu Flüssigkeitsretention und peripheren Ödemen führte und die Hospitalisierungsrate wegen Herzinsuffizienz erhöhte (
94
). Laut der MELODY-1-Studie verbesserte Macitentan PVR, PAWP oder mRAP nach 12-wöchiger Behandlung bei Cpc-PH-Patienten nicht signifikant; der Unterschied war jedoch nicht statistisch signifikant (
95
). Obwohl die Daluxostan-Therapie PAWP, mPAP, PVR oder RAP nicht signifikant senkte, verbesserte sie in der HEAT-Studie den Herzindex bei HF-Patienten nach 3 Wochen. In der Hochdosisgruppe traten Nebenwirkungen häufiger auf. Die EARTH-Studie zeigte auch, dass eine 24-wöchige Behandlung mit Darusentan selbst bei Personen mit echokardiografischem Nachweis von PH den klinischen Zustand oder die kardiale Umgestaltung bei Patienten mit chronischer HF nicht verbesserte. In klinischen Studien zu PH-LHD zeigte ERA generell eine hohe Inzidenz von Nebenwirkungen; mehrere Studien wurden aus Sicherheitsgründen vorzeitig abgebrochen. Daher wird ERA derzeit nicht zur Behandlung von PH-LHD/Cpc-PH empfohlen.Phosphodiesterase (PDE-5-Hemmer)PDE5-Hemmer können die PH und die rechtsventrikuläre Funktion verbessern, indem sie die PDE5-Enzyme hemmen und so zu einer Vasodilatation führen. Ein Cochrane-Review deutet darauf hin, dass die Behandlung mit PDE5-Hemmern die durchschnittliche 6-Minuten-Gehstrecke von Patienten um 48 Meter verlängern kann, während gleichzeitig ihr Funktionsniveau verbessert und das Risiko eines PAH-bedingten Krankenhausaufenthalts gesenkt wird. Die AMBITION-Studie hat gezeigt, dass die anfängliche Kombination von Ambrisentan (ERA) und Tadalafil (PDE5-Hemmer) das Risiko eines klinischen Versagens signifikant senkte. Bei HFrEF-Patienten haben mehrere frühe kleine Studien gezeigt, dass Sildenafil den systolischen pulmonalarteriellen Druck (PASP), mPAP, PVR und 6MWT signifikant senkt und die rechtsventrikuläre Funktion verbessert (
96
,
97
). In einer großen randomisierten kontrollierten Studie ( n = 216) erhöhte Sildenafil im Vergleich zu einer Placebobehandlung weder die körperliche Belastbarkeit, die linksventrikuläre Masse noch kombinierte klinische Endpunkte (Tod, Krankenhausaufenthalt aufgrund von Herz-/Nierenerkrankungen, verstärkte Symptome einer Herzinsuffizienz usw.) . In der Sildenafilgruppe kam es zu einem leichten Anstieg vaskulärer Nebenwirkungen wie Kopfschmerzen, Hitzegefühl und Hypotonie, dieser war jedoch nicht statistisch signifikant (
98
,
99
). Die Wirksamkeit von PDE5-Hemmern bei HFpEF-Patienten ist noch nicht klar, insbesondere bei Patienten ohne rechtsventrikuläre Dysfunktion. Eine rechtsventrikuläre Dysfunktion kann ein wichtiger Prädiktor für den Nutzen einer PDE5-Hemmertherapie sein (
98
). HFpEF-Patienten mit gleichzeitiger Rechtsherzinsuffizienz können von einer PDE5-Hemmertherapie profitieren, da sie beispielsweise den Druck im rechten Vorhof senkt und die Rechtsherzfunktion verbessert. HFpEF-Patienten ohne rechtsventrikuläre Dysfunktion zeigten keinen signifikanten Nutzen (
100
). Mehrere Metaanalysen haben die Wirksamkeit von PDE5-Hemmern (einschließlich Sildenafil) bei PH-LHD zusammengefasst. Bei HFrEF-Patienten verbesserten PDE5-Hemmer den mPAP PVR, die LVEF, die körperliche Leistungsfähigkeit und die Lebensqualität signifikant (
101
,
102
). PDE5-Hemmer wie Sildenafil zeigten bei HFrEF-Patienten bestimmte klinische Vorteile, insbesondere eine Verbesserung der Hämodynamik und der körperlichen Leistungsfähigkeit (
103
). In der SOVIAC-Studie führte Sildenafil bei Patienten mit anhaltender PH nach einer Klappenoperation sogar zu schlechteren klinischen Ergebnissen wie Tod und Krankenhausaufenthalt wegen Herzinsuffizienz (
104
). Die TRITON-Studie untersuchte die Wirksamkeit einer Dreifachkombinationstherapie (Masitentan, Tadalafil und Selexipag) gegenüber einer Zweifachkombinationstherapie (Masitentan und Tadalafil) bei neu diagnostizierten, unbehandelten PH-Patienten, und die Ergebnisse zeigten keinen signifikanten Unterschied beim primären Endpunkt PVR zwischen den beiden. Eine Kohortenanalyse des spanischen PH-Registers untersuchte die prädiktiven Faktoren des Ansprechens auf die Behandlung mit PDE5-Hemmern. Die Ergebnisse zeigten, dass männliches Geschlecht, die Diagnose einer portalen pulmonalen Hypertonie (PoPH) oder HIV-PAH unabhängige Prädiktoren für ein günstiges Ansprechen auf PDE5-Hemmer waren (
105
). Und eine Kohlenmonoxid-Dispersibilität (DLco) ≤ 40 % des vorhergesagten Werts ist mit Nebenwirkungen verbunden. Bei Patienten, die PDE5-Hemmer erhalten haben, aber nicht die gewünschte klinische Wirksamkeit erreicht haben, kann eine Umstellung auf Stimulanzien der löslichen Guanylatcyclase (sGC) in Betracht gezogen werden.Lösliche Guanylcyclase-StimulatorenStimulatoren der löslichen Guanylcyclase fördern die Vasodilatation, indem sie die Produktion von zyklischem Guanosinmonophosphat (cGMP) steigern und so den Mechanismus des PDE5-Hemmers (der durch Verringerung des cGMP-Abbaus wirkt) ergänzen. Sie haben in den letzten Jahren im Bereich der Herzinsuffizienz (HF) und pulmonalen Hypertonie (PH) große Aufmerksamkeit erfahren. In der SOCRATES-REDUCED-Studie wurde Vericiguat von Patienten mit HFrEF gut vertragen, reduzierte jedoch die NT-proBNP-Werte nicht signifikant und verbesserte auch die echokardiographischen Parameter nicht (
106
–
109
). In der SOCRATES-PRESERVED-Studie reduzierte Vericiguat die NT-proBNP-Werte nicht signifikant, war jedoch gut verträglich und mit einer verbesserten Lebensqualität verbunden (
110
). In der VICTORIA-Studie reduzierte Vericiguat den kombinierten Endpunkt aus kardiovaskulärem Tod oder Krankenhauseinweisung wegen Herzinsuffizienz bei Patienten mit sich symptomatischer HFrEF signifikant (
111
). In der LEPHT-Studie wurde Riociguat von Patienten mit HFrEF gut vertragen und verbesserte in der Hochdosisgruppe den Herzindex, den Schlagvolumenindex und den PVR signifikant (
112
). In der DILATE-1-Studie verbesserte Riociguat das Schlagvolumen und den systolischen Blutdruck, hatte jedoch keine signifikante Wirkung auf mPAP und PVR (
113
). In der LEPHT-Studie wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit HFrEF und pulmonaler Hypertonie (PH) untersucht (
114
). Die Studie zeigte, dass die Patienten nach 16-wöchiger Behandlung mit Riociguat (2 mg, dreimal täglich) im Vergleich zur Placebogruppe einen Anstieg des Herzindex und eine signifikante Abnahme des pulmonal-vaskulären Widerstands (PVR) und des systemischen Gefäßwiderstands (SVR) aufwiesen. Die DILATE-1-Studie wurde an Patienten mit HFpEF und PH (LVEF > 50 %, mPAP ≥ 25 mmHg, PAWP > 15 mmHg) durchgeführt. Die Ergebnisse zeigten, dass sich in der Gruppe, die 2 mg Levociguat erhielt, nach 6-stündiger Behandlung das Schlagvolumen und die rechtsventrikuläre enddiastolische Fläche verbesserten, obwohl es zu keiner signifikanten Abnahme des mittleren Pulmonalarteriendrucks (mPAP) kam (
115
). In der Studie COMPERA 2.0 wurden die Sicherheit und Wirksamkeit von Riociguat bei Patienten mit pulmonaler Hypertonie und kardiometabolischen Komorbiditäten untersucht. In der Studie PASSION sollen die Auswirkungen von Riociguat auf die Belastungstoleranz, die Herzfunktionsindizes und die funktionelle WHO-Klassifikation bei Patienten mit Herzinsuffizienz und pulmonaler Hypertonie untersucht werden. In einigen Studien hat Riociguat bei Patienten mit HF-bedingter pulmonaler Hypertonie potenzielle Vorteile gezeigt, wie z. B. verbesserte PVR, SVR und 6MWD, was darauf hindeutet, dass es sich positiv auf die Belastungstoleranz auswirken könnte (
116
). In einer Studie mit 61 Patienten mit pulmonaler Hypertonie (PH), die unzureichend auf PDE-5-Hemmer reagierten, führte die Umstellung auf Riociguat zu einer Erhöhung der 6 MWD nach 24 Wochen, einer Verbesserung des WHO-Grades und einer Senkung der NT-proBNP-Werte (
117
,
118
). Bei Patienten mit Herzinsuffizienz hat Vericiguat bei Patienten mit HFrEF einen signifikanten klinischen Nutzen gezeigt, insbesondere bei der Verringerung des Risikos kardiovaskulärer Ereignisse, hat aber bei Patienten mit HFpEF nur eine begrenzte Wirkung (
119
–
121
). Höhere Dosen von Riociguat verbessern den PVR und den systemischen Gefäßwiderstand bei Patienten mit Cpc-PH, haben aber eine begrenzte Wirksamkeit bei Patienten mit Ipc-PH (
122
,
123
). Die Studie, die Daten aus PATENT-1, PATENT-2, PATENT PLUS und REPLACE umfasste, zeigte, dass Häufigkeit und Schwere der Nebenwirkungen in der Riociguat-Gruppe mit denen in der Placebogruppe vergleichbar waren (
124
).PGI2-MedikamenteProstacyclin wird hauptsächlich von Endothelzellen produziert und hat gefäßerweiternde, antithrombotische und antiproliferative Wirkungen, die die Hämodynamik und die Herzfunktion verbessern können. Zu den Prostacyclin-Medikamenten gehören Prostacyclin-Analoga (wie Epoprostenol, Treprostinil, Iloprost, Beraprost) und Prostacyclin-IP-Rezeptoragonisten (wie Selexipag). Eprostol wirkt, indem es den PVR senkt und die Funktion des rechten Ventrikels verbessert, aber die langfristige Einnahme kann schädliche neurohormonale Systeme (wie das Renin-Angiotensin-System) aktivieren und so eine Herzinsuffizienz verschlimmern. Obwohl Epprostol in der FIRST-Studie den Herzindex, den pulmonalen Verschlussdruck (PAWP) und den systemischen Gefäßwiderstand (SVR) signifikant verbesserte, wurde die Studie aufgrund der verringerten Patientenüberlebensrate vorzeitig abgebrochen. Treprolinil ist ein neuartiges Prostacyclin-Analogon, das den Stoffwechsel und die Herzfunktion verbessern kann, indem es den AMPK-Signalweg in der Skelettmuskulatur und dem rechten Ventrikel aktiviert. In Tiermodellen verbesserte Trepronnier das metabolische Syndrom und senkte den pulmonalarteriellen Druck und zeigte damit das Potenzial, die Entwicklung einer Herzinsuffizienz (HFpEF) mit Ejakulationsfraktionen zu verhindern. Anders als eine Herzinsuffizienz (HFrEF), die eine reduzierte Auswurffraktion aufweist, wird HFpEF häufig von einem metabolischen Syndrom und Diabetes begleitet, und die günstigen Wirkungen von Prostacyclin-Analoga auf den Stoffwechsel und die Lungenblutgefäße machen es möglich, dass es für die Behandlung von PH-HFpEF besser geeignet ist. Bei Patienten mit Cpc-PH verbessert eine gezielte Therapie der pulmonalen Hypertonie (wie Phosphodiesterase-5-Hemmer und Endothelin-Rezeptor-Antagonisten) die Symptome nicht, sondern erhöht stattdessen die Morbiditäts- und Mortalitätsrate. Eine Metaanalyse hat gezeigt, dass eine auf PH ausgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko unerwünschter Ereignisse jedoch höher ist. Eine Metaanalyse hat gezeigt, dass eine pH-zielgerichtete Therapie zwar die körperliche Leistungsfähigkeit von Patienten mit Linksherzerkrankung (LHD) verbessern kann, das Risiko für Nebenwirkungen jedoch höher ist. Die Wirksamkeit bei menschlichen pH-HFPEF-Patienten muss jedoch noch durch weitere klinische Studien bestätigt werden. Zudem könnte die Behandlungsstrategie der Skelettmuskulatur und des rechtsventrikulären AMPK-Signalwegs eine mögliche Richtung für die Prävention von PH-LHD sein (
125
–
131
).Ralinepag, ein neuartiger Prostazyklin-Rezeptor (IP-Rezeptor)-Agonist, zeigt signifikante gefäßerweiternde Wirkungen, die PVR wirksam reduzieren und gleichzeitig die Proliferation glatter Gefäßmuskelzellen sowie die Thrombozytenaggregation hemmen können. Das Medikament hat eine Halbwertszeit von bis zu 24 Stunden, was ihm einen potenziellen Vorteil bei der Behandlung von PH verschafft. In klinischen Studien der Phase II hat Ralinepag eine gute Wirksamkeit gezeigt. Zu den häufigen Nebenwirkungen zählen Kopfschmerzen, Übelkeit und Durchfall. Derzeit wird die klinische Studie der Phase III zu Ralinepag vorangetrieben, und die vorläufigen Langzeitdaten der ADVANCE EXTENSION-Studie wurden auf entsprechenden wissenschaftlichen Konferenzen veröffentlicht (
132
). Derzeit ist die breite Anwendung von Prostazyklin-Medikamenten in der klinischen Praxis aufgrund ihrer geringen Stabilität und vieler Nebenwirkungen begrenzt. Daher ist die Entwicklung neuartiger Prostazyklin-Medikamente mit höherer Rezeptorselektivität, weniger Nebenwirkungen, höherer Stabilität, einfacherer Konservierung und höherer Akzeptanz bei den Patienten zur Forschungsrichtung geworden (
133
).SotatercepSotatercept ist ein Fusionsprotein des Aktivinrezeptors Typ IIA (ActRIIA). Ergebnisse der PULSAR-Studie zeigen, dass Sotatercept Placebo bei der Senkung des PVR und der Verbesserung der 6MWD sowie bei sekundären Endpunkten wie NT-proBNP-Spiegel, funktioneller WHO-Klassifikation und klinischer Verschlechterung deutlich übertrifft. Ergebnisse der STELLAR-Studie zeigen, dass Patienten in der Sotatercept-Gruppe nach 24-wöchiger Behandlung eine Steigerung der 6MWD um 40,8 Meter verzeichneten, verglichen mit einer Steigerung von lediglich 1,0 Meter in der Placebogruppe. Andere sekundäre Endpunkte, darunter Senkungen der NT-proBNP-Spiegel, Verbesserungen der funktionellen WHO-Klassifikation und Verringerung des PVR, waren in der Sotatercept-Gruppe ebenfalls signifikant besser. Bei einer medianen Nachbeobachtungszeit von 32,7 Wochen reduzierte Sotatercept das Risiko einer klinischen Verschlechterung oder des Todes um 84 %. Darüber hinaus verbesserte Sotatercept die Rechtsherzfunktion durch Senkung des mPAP und Verringerung der Arbeitsbelastung der rechten Herzseite. In Bezug auf die Sicherheit umfassten die Nebenwirkungen in der Sotatercept-Gruppe hauptsächlich Kapillarerweiterung, Nasenbluten, Zahnfleischbluten und Thrombozytopenie, die meisten dieser Ereignisse waren jedoch mild. In der Post-hoc- Analyse der STELLAR-Studie wurde festgestellt, dass Sotatercept die Größe der rechten Herzseite reduzierte und die rechtsventrikuläre Funktion sowie die hämodynamischen Parameter nach 24-wöchiger Behandlung verbesserte. Laufende Studien wie SOTERIA, HYPERION, ZENITH, CADENCE, MK-7962-020 und MOONBEAM werden die langfristige Sicherheit und Wirksamkeit von Sotatercept in verschiedenen Patientenpopulationen weiter erforschen und umfassendere klinische Belege für die künftige Behandlung der pulmonalen arteriellen Hypertonie liefern. Im Vergleich zu herkömmlichen niedermolekularen Verbindungen hat Sotatercept eine höhere Wirksamkeit und weniger Nebenwirkungen. Durch die Wiederherstellung des Gleichgewichts des BMPR2-Signalwegs und die Hemmung des Smad2/3-Signalwegs reduziert Sotatercept die Proliferation glatter Lungengefäßmuskelzellen und die Kollagenablagerung und kehrt so den Lungengefäßumbau um. Dieses Ziel ist mit den meisten derzeit verfügbaren zielgerichteten Medikamenten nicht erreichbar. Darüber hinaus sind das Dosierungsschema von Sotatercept von einmal alle drei Wochen und die subkutane Injektionsmethode bequemer als bei herkömmlichen Medikamenten (
Tabelle 4
). Mit seinem einzigartigen Wirkmechanismus und den erheblichen klinischen Vorteilen bietet Sotatercept eine neue Behandlungsoption für Cpc-PH-Patienten und hat das Potenzial, die derzeitige Behandlungslandschaft zu verändern (
134
–
138
). Tabelle 4
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.