
- Beiträge: 1755
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Chronische thromboembolische pulmonale Hypertonie: die therapeutische Bewertung
Chronische thromboembolische pulmonale Hypertonie: die therapeutische Bewertung
16 Dez 2024 14:29 #2299
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Chronische thromboembolische pulmonale Hypertonie: die therapeutische Bewertung wurde erstellt von danny
www.frontiersin.org/journals/cardiovascu...vm.2024.1439411/full
Chronische thromboembolische pulmonale Hypertonie: die therapeutische Bewertung
Chronische thromboembolische pulmonale Hypertonie (CTEPH) ist eine schwere und komplexe Erkrankung, die sich aus einer nicht behandelten Lungenembolie entwickelt und zu einer fibrotischen Obstruktion der Lungenarterien, pulmonaler Hypertonie und potenzieller Rechtsherzinsuffizienz führt. Der Eckpfeiler der CTEPH-Behandlung ist ein vielschichtiger therapeutischer Ansatz, der auf das individuelle Patientenprofil zugeschnitten ist und die Heterogenität der Krankheit widerspiegelt. Dieser Bericht befasst sich mit den aktuellen Therapiestrategien für CTEPH, darunter chirurgische Lungenendarteriektomie (PEA), Ballon-Pulmonalangioplastie (BPA) und gezielte pharmakologische Behandlungen wie PDE5-Hemmer, Endothelin-Rezeptor-Antagonisten, sGC-Stimulatoren und Prostanoide. Eine lebenslange Antikoagulation wird ebenfalls als vorbeugende Strategie gegen wiederkehrende Thromboembolien hervorgehoben. Besonderer Wert wird auf die interdisziplinäre Natur der CTEPH-Behandlung gelegt, die eine Zusammenarbeit zwischen PEA-Chirurgen, BPA-Interventionisten, PH-Spezialisten und Thoraxradiologen erfordert, um eine umfassende Behandlungsplanung und -durchführung sicherzustellen. Die Überprüfung unterstreicht die Bedeutung der Auswahl einer geeigneten Behandlungsmethode auf der Grundlage der spezifischen Krankheitsmerkmale des Patienten und der sich entwickelnden Landschaft der CTEPH-Behandlung, mit dem Ziel, die Patientenergebnisse durch integrierte Behandlungsstrategien zu verbessern. 1 EinleitungPulmonale Hypertonie (PH) ist definiert als ein mittlerer pulmonalarterieller Druck (PAP) von größer oder gleich 20 mmHg, gemessen durch Rechtsherzkatheterisierung (RHC) ( 1 , 2 ).Die chronisch thromboembolische Lungenerkrankung (CTEPH), eine potenziell lebensbedrohliche Erkrankung, ist definiert als symptomatische pulmonale Hypertonie mit anhaltenden Lungendurchblutungsstörungen trotz adäquater Antikoagulation über 3–6 Monate ( 3 ) und stellt eine eigenständige Krankheitseinheit dar, die gemäß den ESC/ERS-Leitlinien 2022 ( 2 ) als pulmonale Hypertonie (PH) der Gruppe 4 klassifiziert wird .CTEPH ist eine seltene und unterdiagnostizierte Komplikation einer akuten Lungenembolie (APE) ( 4 ). Einige Merkmale der ursprünglichen Lungenembolie sind mit der Entwicklung von CTEPH assoziiert. Besonders wichtig ist, dass eine unprovozierte Lungenembolie, eine Diagnoseverzögerung von >2 Wochen und eine Funktionsstörung des rechten Ventrikels (RV) zum Zeitpunkt der Lungenembolie unabhängige Prädiktoren für CTEPH sind ( 5 ).Darüber hinaus ist die genaue Inzidenz von CTEPH nach einer dokumentierten und korrekt behandelten APE noch immer unbekannt.Die genaue Epidemiologie der CTEPH ist unbekannt; sie wird höchstwahrscheinlich weitgehend unterdiagnostiziert und daher unterbehandelt und zeigt, wie sehr Patienten von modernen multimodalen Behandlungskonzepten in Expertenzentren profitieren können.Chronische thromboembolische Lungenerkrankung (CTEPD) ist ein allgemeiner Begriff, der im jüngsten ERS-Statement zur chronischen thromboembolischen pulmonalen Hypertonie (CTEPH) ( 6 ) vorgeschlagen wurde, um symptomatische Patienten zu charakterisieren, die nach mindestens drei Monaten therapeutischer Antikoagulation in der Ventilations-/Perfusions-Lungenszintigraphie (V/Q) nicht übereinstimmende Perfusionsdefekte und in der Computertomographie-Pulmonalisangiographie (CTPA), konventionellen Pulmonalisangiographie (CPA) oder Magnetresonanztomographie (MRT) spezifische Anzeichen chronischer organisierter Gerinnsel aufweisen. Einige dieser Patienten weisen im Ruhezustand keine pulmonale Hypertonie (PH) auf ( 2 ), während bei der Mehrheit eine PH im Ruhezustand vorliegt, entsprechend der Definition der CTEPH (Gruppe 4 der aktualisierten klinischen Klassifikation der PH) ( 1 ).Der natürliche Verlauf von CTEPH ist komplex. Das aktuelle Verständnis der Pathophysiologie von CTEPH hat sich in letzter Zeit über eine einfache pulmonalvaskuläre Erkrankung hinaus entwickelt, die durch eine intravaskuläre mechanische Obstruktion aufgrund von organisiertem chronischem fibrotischem Material aus nicht aufgelösten Blutgerinnseln verursacht wird. Tatsächlich könnte es sich um eine viel komplexere Erkrankung handeln, die eine proximale und distalere Obstruktion der Lungenarterien umfasst, die mit einer Umgestaltung der muskulären Lungenarterien, Kapillaren und Venolen einhergeht ( 7 ).Heute ist es allgemein anerkannt, dass pulmonale Gefäßumbauprozesse zu einer signifikanten pulmonalen Mikrovaskulopathie führen können, die wiederum bei der Entwicklung eines progressiven Anstiegs des pulmonalen Gefäßwiderstands und in der Folge bei der Entstehung von Funktionsstörungen der rechten Herzhälfte und symptomatischer CTEPH eine Rolle spielt ( 8 , 9 ). Die Prozesse, die zur Persistenz organisierter Gerinnsel nach einer APE führen, sind noch nicht vollständig verstanden, auch wenn mehrere Risikofaktoren identifiziert wurden.Die Risikofaktoren für CTEPH scheinen sich von denen für APE zu unterscheiden, wie z. B. Immobilisierung oder eine kürzlich erfolgte Operation. Zu den potenziellen Risikofaktoren für CTEPH gehören bestimmte chronische Erkrankungen (z. B. permanente intravaskuläre Geräte, entzündliche Darmerkrankungen, Autoimmunerkrankungen, Hypothyreose, Splenektomie und Malignität), Thrombophilie und genetische Veranlagung.Zur Diagnostik können Echokardiographie, CT und Ventilations-Perfusions-Scans (V/Q-Scans) eingesetzt werden, um den Verdacht auf PH zu bestätigen. Das Vorliegen einer V/Q-Inkongruenz im Rahmen einer PH sollte eine weitere Untersuchung mittels RHC und Pulmonalisangiographie nach sich ziehen. Jedes Bildgebungsverfahren hat seine Funktion; daher ist eine umfassende Untersuchung mittels multimodaler Bildgebung für die richtige Diagnose und Behandlung von Patienten mit CTEPH entscheidend. Die Seltenheit der Erkrankung, unspezifische Symptome und Anzeichen sowie die mangelnde Kenntnis der Ärzte über CTEPH (einschließlich des Zeitpunkts des Verdachts und der Art und Weise, wie sie zu beurteilen ist) erschweren eine rechtzeitige Diagnose ( 10 ).Daher werden viele Patienten trotz wirksamer Therapie [pulmonale Endarteriektomie (PEA)] erst im Spätstadium der Erkrankung diagnostiziert, wenn die distale PA-Obstruktion und die Mikrovaskulopathie bereits fortgeschritten sind. Patienten mit fortgeschrittener Erkrankung kommen für eine PEA nicht in Frage, da mit diesem Verfahren nur proximale Läsionen behandelt werden können.Glücklicherweise wurden parallel zu den Fortschritten bei den Diagnose- und Operationstechniken für Patienten mit operablem CTEPH auch erhebliche therapeutische Fortschritte eingeführt. Dazu gehören die gezielte medizinische Therapie und die Ballon-Pulmonalisangioplastie (BPA) für Patienten, bei denen aufgrund der distalen Lage der chronischen thromboembolischen Obstruktion, der Schwere der hämodynamischen Beeinträchtigung, des Vorhandenseins schwerer Komorbiditäten oder persönlicher Präferenz eine inoperable CTEPH angenommen wird ( 11 – 14 ).Daher sind für optimale Patientenergebnisse ein hohes Verdachtspotenzial und eine rechtzeitige Diagnose mithilfe multimodaler Bildgebungsverfahren zwingend erforderlich.Ziel der vorliegenden Übersicht ist es, das Bewusstsein für diese seltene Erkrankung zu schärfen und neuere Bemühungen zur Entwicklung von Behandlungslösungen für verschiedene Phänotypen der CTEPH darzulegen.
2 TherapieDie moderne Therapiestrategie für chronisch thromboembolische pulmonale Hypertonie (CTEPH) verfolgt einen umfassenden, multimodalen Ansatz, der pulmonale Endarteriektomie (PEA), pulmonale Ballonangioplastie (BPA) und pharmakologische Behandlungen integriert, um die vielfältigen anatomischen Herausforderungen durch proximale, distale und mikrovaskuläre Läsionen zu bewältigen ( Abbildung 1 ). PEA stellt eine potenziell kurative Option dar, ihre Anwendbarkeit ist jedoch durch das Operationsrisiko oder die Unzugänglichkeit der Krankheit bei bestimmten Patienten beschränkt, sodass alternative Eingriffe wie BPA und medikamentöse Therapie erforderlich sind. In Ausnahmefällen, etwa bei Patienten mit gemischten anatomischen Läsionen (operativ erreichbare Läsionen in einer Lunge und inoperable Läsionen in der anderen Lunge), kann ein kombinierter Ansatz mit BPA (vor oder gleichzeitig mit der Operation) und PEA von Nutzen sein, um das Operationsrisiko zu senken und das Endergebnis zu verbessern ( 2 ). Abildung 1
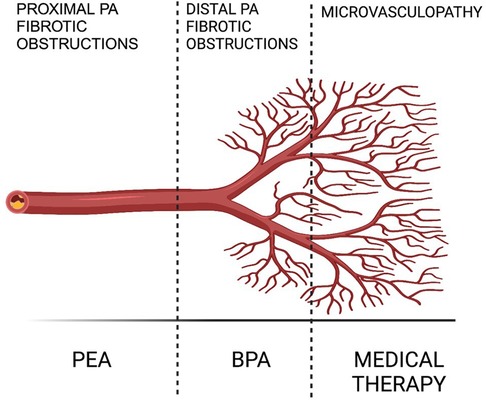 Abbildung 1. Multimodale CTEPH-Behandlung (
2
). 2.1 Chirurgische BehandlungEine genaue Diagnose und eine sorgfältige Patientenauswahl sind entscheidend für die Eignung einer pulmonalen Endarteriektomie (PEA) zur Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH). Kandidaten für diesen chirurgischen Eingriff sollten eine Dyspnoe bei Belastung aufweisen, die mindestens der NYHA-Funktionsklasse II entspricht. Das obstruktive Material muss auf Computertomographie-Scans (CT) erkennbar und als chirurgisch zugänglich gelten. Typischerweise umfasst es zentrale, segmentale und subsegmentale Pulmonalarterien (
15
).Die Indikation zur Operation bei CTEPH-Patienten hängt von mehreren Faktoren ab: Schwere der Symptome, Grad der PH und Rechtsherzfunktionsstörung, Grad der Stenosen und Verschlüsse, Zusammenhang zwischen PH und Ausmaß der Obstruktionen, Komorbiditäten und mögliche Operationsschwierigkeiten, Erwartungen des Patienten und Risikobereitschaft.Jüngste Erkenntnisse legen nahe, dass eine PEA bei einem erheblichen Teil der CTEPH-Patienten durchführbar ist, insbesondere wenn die Entscheidung auf der Evaluierung durch ein interdisziplinäres Team beruht (
16
,
17
). Weder Alter noch Komorbiditäten gelten als absolute Kontraindikationen für eine PEA. Mögliche Risikofaktoren, die sich auf das Operationsergebnis und die allgemeine Lebenserwartung auswirken, müssen jedoch im Entscheidungsprozess sorgfältig abgewogen werden.Die Kriterien für die Auswahl von Patienten für PEA sind in
Tabelle 1
detailliert aufgeführt (
18
–
20
). Tbelle 1
Abbildung 1. Multimodale CTEPH-Behandlung (
2
). 2.1 Chirurgische BehandlungEine genaue Diagnose und eine sorgfältige Patientenauswahl sind entscheidend für die Eignung einer pulmonalen Endarteriektomie (PEA) zur Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH). Kandidaten für diesen chirurgischen Eingriff sollten eine Dyspnoe bei Belastung aufweisen, die mindestens der NYHA-Funktionsklasse II entspricht. Das obstruktive Material muss auf Computertomographie-Scans (CT) erkennbar und als chirurgisch zugänglich gelten. Typischerweise umfasst es zentrale, segmentale und subsegmentale Pulmonalarterien (
15
).Die Indikation zur Operation bei CTEPH-Patienten hängt von mehreren Faktoren ab: Schwere der Symptome, Grad der PH und Rechtsherzfunktionsstörung, Grad der Stenosen und Verschlüsse, Zusammenhang zwischen PH und Ausmaß der Obstruktionen, Komorbiditäten und mögliche Operationsschwierigkeiten, Erwartungen des Patienten und Risikobereitschaft.Jüngste Erkenntnisse legen nahe, dass eine PEA bei einem erheblichen Teil der CTEPH-Patienten durchführbar ist, insbesondere wenn die Entscheidung auf der Evaluierung durch ein interdisziplinäres Team beruht (
16
,
17
). Weder Alter noch Komorbiditäten gelten als absolute Kontraindikationen für eine PEA. Mögliche Risikofaktoren, die sich auf das Operationsergebnis und die allgemeine Lebenserwartung auswirken, müssen jedoch im Entscheidungsprozess sorgfältig abgewogen werden.Die Kriterien für die Auswahl von Patienten für PEA sind in
Tabelle 1
detailliert aufgeführt (
18
–
20
). Tbelle 1
 Tabelle 1. Indikationskriterien für PEA. Obwohl Chirurgen bei einem sehr hohen PVR (insbesondere > 1.100 dyn) Bedenken äußern, da dies auf eine stärker distale Erkrankung hindeuten kann, besagen aktuelle Empfehlungen, dass ein erhöhter PVR (> 1.500 dyn·s·cm −5 ) allein keine Kontraindikation für eine Operation darstellt. Tatsächlich gibt es keine PVR-Obergrenze, ab der ein Patient inoperabel ist, vorausgesetzt, es liegt ein entsprechender Grad der obstruktiven Erkrankung vor. Bei einigen Patienten kann jedoch ein stark erhöhter PVR in Kombination mit anderen Risikofaktoren eine Operation unmöglich machen. Darüber hinaus entscheiden sich manche Patienten mit operablem Zustand möglicherweise gegen eine Operation.Ein wichtiger Aspekt ist die technische Zugänglichkeit des obstruktiven Materials. Diese hängt in erster Linie von der chirurgischen Expertise ab, die wiederum mit der Fallzahl des einzelnen Chirurgen korreliert. Das Operationsrisiko hängt vom Schweregrad der hämodynamischen Beeinträchtigung im Verhältnis zur Menge des obstruktiven Materials, von Komorbiditäten und vom Alter der Patienten ab (
20
).In erster Linie wird eine Operation bei Patienten mit CTEPH vom zentralen Typ empfohlen, die durch Verstopfungen der Haupt-, Interlobär- und Segmentpulmonalarterien sowie Wandthromben und Intimahyperplasie gekennzeichnet ist. Leider sind Patienten mit peripherer CTEPH, bei denen die Krankheit auch weiter entfernte Pulmonalarterien befällt, möglicherweise keine geeigneten Kandidaten für einen effektiven chirurgischen Eingriff.Ein bedeutender chirurgischer Fortschritt in der Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) besteht in der Erweiterung der distalen Grenzen der Endarteriektomie, wodurch in Expertenzentren sogar bei Fällen distaler chronischer Thromboembolie erfolgreiche Operationen möglich sind (
20
,
21
). Dieser Fortschritt ist größtenteils den verbesserten Diagnosemöglichkeiten und der erworbenen chirurgischen Expertise zuzuschreiben. Aus diesem Grund hat die University of California in San Diego ihre zuvor veröffentlichte intraoperative Klassifikation überarbeitet, um die moderne Operationsmethodik und das Ausmaß der erreichten Revaskularisierung genauer darzustellen (
Tabelle 2
) (
20
,
22
,
23
). Tbelle 2
Tabelle 1. Indikationskriterien für PEA. Obwohl Chirurgen bei einem sehr hohen PVR (insbesondere > 1.100 dyn) Bedenken äußern, da dies auf eine stärker distale Erkrankung hindeuten kann, besagen aktuelle Empfehlungen, dass ein erhöhter PVR (> 1.500 dyn·s·cm −5 ) allein keine Kontraindikation für eine Operation darstellt. Tatsächlich gibt es keine PVR-Obergrenze, ab der ein Patient inoperabel ist, vorausgesetzt, es liegt ein entsprechender Grad der obstruktiven Erkrankung vor. Bei einigen Patienten kann jedoch ein stark erhöhter PVR in Kombination mit anderen Risikofaktoren eine Operation unmöglich machen. Darüber hinaus entscheiden sich manche Patienten mit operablem Zustand möglicherweise gegen eine Operation.Ein wichtiger Aspekt ist die technische Zugänglichkeit des obstruktiven Materials. Diese hängt in erster Linie von der chirurgischen Expertise ab, die wiederum mit der Fallzahl des einzelnen Chirurgen korreliert. Das Operationsrisiko hängt vom Schweregrad der hämodynamischen Beeinträchtigung im Verhältnis zur Menge des obstruktiven Materials, von Komorbiditäten und vom Alter der Patienten ab (
20
).In erster Linie wird eine Operation bei Patienten mit CTEPH vom zentralen Typ empfohlen, die durch Verstopfungen der Haupt-, Interlobär- und Segmentpulmonalarterien sowie Wandthromben und Intimahyperplasie gekennzeichnet ist. Leider sind Patienten mit peripherer CTEPH, bei denen die Krankheit auch weiter entfernte Pulmonalarterien befällt, möglicherweise keine geeigneten Kandidaten für einen effektiven chirurgischen Eingriff.Ein bedeutender chirurgischer Fortschritt in der Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) besteht in der Erweiterung der distalen Grenzen der Endarteriektomie, wodurch in Expertenzentren sogar bei Fällen distaler chronischer Thromboembolie erfolgreiche Operationen möglich sind (
20
,
21
). Dieser Fortschritt ist größtenteils den verbesserten Diagnosemöglichkeiten und der erworbenen chirurgischen Expertise zuzuschreiben. Aus diesem Grund hat die University of California in San Diego ihre zuvor veröffentlichte intraoperative Klassifikation überarbeitet, um die moderne Operationsmethodik und das Ausmaß der erreichten Revaskularisierung genauer darzustellen (
Tabelle 2
) (
20
,
22
,
23
). Tbelle 2
 Tabelle 2. Morphologie der PA-Okklusion (chirurgische Klassifikation chronischer Thromboembolien der University of California San Diego). Chronische thromboembolische Erkrankungen (CTEPD) stellen eine einzigartige Untergruppe von Erkrankungen dar, die hinsichtlich Symptomen und Durchblutungsstörungen der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) stark ähneln, aber deutlich von pulmonaler Hypertonie (PH) im Ruhezustand abweichen. Diese Erkrankung umfasst die gleichen morphologischen Anzeichen wie CTEPH, z. B. V/Q-Missverhältnis und trotz Antikoagulationstherapie anhaltende klinische Symptome, ohne dass die formalen PH-Kriterien erfüllt sind [mittlerer pulmonalarterieller Druck (mPAP) < 20 mmHg]. In Fällen, in denen ein erhebliches V/Q-Missverhältnis vorliegt und eine Linderung der Symptome zu erwarten ist, kann ein chirurgischer Eingriff in Betracht gezogen werden. Die Entscheidung, eine pulmonale Endarteriektomie (PEA) durchzuführen, hängt nicht allein vom Grad des pulmonalvaskulären Widerstands (PVR) oder der rechtsventrikulären Dysfunktion ab, da keines von beiden als absolute Kontraindikationen gilt. Die Entscheidung über die operative Eignung einer PEA erfolgt individuell und interdisziplinär unter Berücksichtigung verschiedener, in speziellen Leitlinien beschriebener Faktoren und sollte immer an einem auf diese Behandlungen spezialisierten Zentrum erfolgen (
Tabelle 3
) (
15
,
19
). Tbelle 3
Tabelle 2. Morphologie der PA-Okklusion (chirurgische Klassifikation chronischer Thromboembolien der University of California San Diego). Chronische thromboembolische Erkrankungen (CTEPD) stellen eine einzigartige Untergruppe von Erkrankungen dar, die hinsichtlich Symptomen und Durchblutungsstörungen der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) stark ähneln, aber deutlich von pulmonaler Hypertonie (PH) im Ruhezustand abweichen. Diese Erkrankung umfasst die gleichen morphologischen Anzeichen wie CTEPH, z. B. V/Q-Missverhältnis und trotz Antikoagulationstherapie anhaltende klinische Symptome, ohne dass die formalen PH-Kriterien erfüllt sind [mittlerer pulmonalarterieller Druck (mPAP) < 20 mmHg]. In Fällen, in denen ein erhebliches V/Q-Missverhältnis vorliegt und eine Linderung der Symptome zu erwarten ist, kann ein chirurgischer Eingriff in Betracht gezogen werden. Die Entscheidung, eine pulmonale Endarteriektomie (PEA) durchzuführen, hängt nicht allein vom Grad des pulmonalvaskulären Widerstands (PVR) oder der rechtsventrikulären Dysfunktion ab, da keines von beiden als absolute Kontraindikationen gilt. Die Entscheidung über die operative Eignung einer PEA erfolgt individuell und interdisziplinär unter Berücksichtigung verschiedener, in speziellen Leitlinien beschriebener Faktoren und sollte immer an einem auf diese Behandlungen spezialisierten Zentrum erfolgen (
Tabelle 3
) (
15
,
19
). Tbelle 3
 Tabelle 3. Zusammenfassung der wichtigsten Faktoren, die die therapeutische Indikation für PEA beeinflussen. Bei Patienten mit Kontraindikationen oder relativen Kontraindikationen für einen kardiopulmonalen Bypass ist hinsichtlich einer pulmonalen Endarteriektomie (PEA) Vorsicht geboten, da bei diesem Verfahren ein kardiopulmonaler Bypass über eine Kanülierung der Aorta ascendens und der A. cava erforderlich ist, was zu Phasen eines tiefen hypothermischen Kreislaufstillstands (HCA) führen kann. Hochrisikopatienten oder solche mit Erkrankungen wie neurologischen Erkrankungen, kürzlich erlittenen Schlaganfällen, Herzinfarkten, Blutungsstörungen, schwerer Linksherzinsuffizienz, aktiven Infektionen, anderen Lungenerkrankungen, bösartigen Tumoren, kürzlich erlittenem Lungentrauma und Multiorganversagen profitieren möglicherweise nicht von einer PEA (
24
).Die PEA wird über eine mediane Sternotomie durchgeführt, wobei der Patient an eine Herz-Lungen-Maschine angeschlossen und der Körper auf 18–20 °C gekühlt wird, um eine vollständige HCA zu induzieren. Dieser Zustand ist wichtig, um das obstruktive thrombotische Material aus der Intima und der inneren Tunica media unter optimaler Sicht und ohne Rückfluss des Kollateralkreislaufs zu entfernen. Kurze Kreislaufstillstände, typischerweise 20 min mit dazwischenliegenden Reperfusionsintervallen, werden eingelegt, um das Risiko für die dünnen Arterienwände zu minimieren und gleichzeitig eine gründliche Entfernung des Materials in die distalen Gefäße sicherzustellen. Komorbide Herzerkrankungen können während dieser Zeit ebenfalls behandelt werden, wie z. B. ein Koronararterien-Bypass (CABG), der Verschluss eines offenen formanen ovalis (PFO)/Vorhofseptumdefekts (ASD) oder eine partielle Korrektur einer anomalen Pulmonalvene (
19
).Die Komplexität des Verfahrens trägt zu einem breiten Spektrum potenzieller Komplikationen bei, darunter Arrhythmien, postoperatives Rechtsventrikelversagen (RV) aufgrund von Resterkrankung und anhaltender PH, Reperfusionslungenödem und Lungenverletzung, anhaltende Hypoxämie und das Risiko einer Arteriotomieruptur oder distalen Arterienperforation, die zu massiver Hämoptyse führt (
25
). Frühe Komplikationen wie ein Reperfusionslungenödem, das sich als erhöhter Sauerstoffbedarf und Lungentrübung manifestiert, können eine längere Intubation oder VA-ECMO-Unterstützung erforderlich machen. Eine Funktionsstörung des rechten Ventrikels kann neben anderen Komplikationen wie nosokomialen Pneumonien auch zum Einsatz von ECMO führen. Eine erneute Thrombose in endarteriektomierten Bereichen ist selten, und obwohl HCA die Wachsamkeit vorübergehend verringern kann, wurde sein Einsatz nicht mit anhaltenden kognitiven Dysfunktionen in Verbindung gebracht (
17
,
19
,
26
).Bei unbehandelter CTEPH liegt die 3-Jahres-Überlebensrate bei 70 %, wobei dies je nach hämodynamischer Schwere variiert (
17
,
27
). Durch PEA wird die Prognose signifikant verbessert, die 3-Jahres-Überlebensrate beträgt 89 % (
17
). Die chirurgische Sterblichkeitsrate hat sich drastisch von >20 % auf <5 % reduziert und ist nun mit anderen Herzoperationen vergleichbar (
26
,
28
,
29
). Nach PEA zeigen sich sofortige hämodynamische Verbesserungen, darunter ein verringerter pulmonalarterieller Druck (PAP), wobei innerhalb von drei bis zwölf Monaten eine Umgestaltung des rechten Ventrikels und eine Leistungssteigerung auftreten. Ein Jahr nach PEA sind eine signifikante Verringerung des pulmonalvaskulären Widerstands (PVR), Verbesserungen der 6-Minuten-Gehstrecke und Verbesserungen der NYHA-Funktionsklasse zu beobachten. 17–51 % der Patienten leiden an einer anhaltenden PH, häufig aufgrund von Mikrovaskularerkrankungen, technischen Herausforderungen bei der Entfernung distalen Materials oder Komorbiditäten. Dennoch erfahren diese Patienten im Allgemeinen eine klinische Besserung (
20
,
24
).2.2 Interventionelle Behandlung: Ballon-Pulmonalisangioplastie (BPA)Die pulmonale Ballonangioplastie (BPA) ist ein interventionelles Verfahren, das in mehreren Sitzungen durchgeführt wird und sich besonders zur Behandlung distaler subsegmentaler pulmonalarterieller Obstruktionen eignet, die sich einem chirurgischen Eingriff nicht unterziehen lassen. Kurzfristig hat die BPA im Vergleich zur Pharmakotherapie eine bessere Wirkung auf die Hämodynamik gezeigt, darunter eine Verbesserung der 6-Minuten-Gehstrecke, des pulmonalvaskulären Widerstands (PVR) und des mittleren pulmonalarteriellen Drucks (mPAP) (
15
,
30
). Daher ist die BPA typischerweise Patienten mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) vorbehalten, die keine Kandidaten für eine pulmonale Endarteriektomie (PEA) sind, oder die nach der Operation eine Rest-PH aufweisen (
31
). Die primäre Indikation für die BPA besteht in Szenarien, in denen PEA keine Option ist und sich Medikamente als unwirksam erwiesen haben. Für ihre Durchführung sind bestimmte Kriterien festgelegt (
Tabelle 4
). Tbelle 4
Tabelle 3. Zusammenfassung der wichtigsten Faktoren, die die therapeutische Indikation für PEA beeinflussen. Bei Patienten mit Kontraindikationen oder relativen Kontraindikationen für einen kardiopulmonalen Bypass ist hinsichtlich einer pulmonalen Endarteriektomie (PEA) Vorsicht geboten, da bei diesem Verfahren ein kardiopulmonaler Bypass über eine Kanülierung der Aorta ascendens und der A. cava erforderlich ist, was zu Phasen eines tiefen hypothermischen Kreislaufstillstands (HCA) führen kann. Hochrisikopatienten oder solche mit Erkrankungen wie neurologischen Erkrankungen, kürzlich erlittenen Schlaganfällen, Herzinfarkten, Blutungsstörungen, schwerer Linksherzinsuffizienz, aktiven Infektionen, anderen Lungenerkrankungen, bösartigen Tumoren, kürzlich erlittenem Lungentrauma und Multiorganversagen profitieren möglicherweise nicht von einer PEA (
24
).Die PEA wird über eine mediane Sternotomie durchgeführt, wobei der Patient an eine Herz-Lungen-Maschine angeschlossen und der Körper auf 18–20 °C gekühlt wird, um eine vollständige HCA zu induzieren. Dieser Zustand ist wichtig, um das obstruktive thrombotische Material aus der Intima und der inneren Tunica media unter optimaler Sicht und ohne Rückfluss des Kollateralkreislaufs zu entfernen. Kurze Kreislaufstillstände, typischerweise 20 min mit dazwischenliegenden Reperfusionsintervallen, werden eingelegt, um das Risiko für die dünnen Arterienwände zu minimieren und gleichzeitig eine gründliche Entfernung des Materials in die distalen Gefäße sicherzustellen. Komorbide Herzerkrankungen können während dieser Zeit ebenfalls behandelt werden, wie z. B. ein Koronararterien-Bypass (CABG), der Verschluss eines offenen formanen ovalis (PFO)/Vorhofseptumdefekts (ASD) oder eine partielle Korrektur einer anomalen Pulmonalvene (
19
).Die Komplexität des Verfahrens trägt zu einem breiten Spektrum potenzieller Komplikationen bei, darunter Arrhythmien, postoperatives Rechtsventrikelversagen (RV) aufgrund von Resterkrankung und anhaltender PH, Reperfusionslungenödem und Lungenverletzung, anhaltende Hypoxämie und das Risiko einer Arteriotomieruptur oder distalen Arterienperforation, die zu massiver Hämoptyse führt (
25
). Frühe Komplikationen wie ein Reperfusionslungenödem, das sich als erhöhter Sauerstoffbedarf und Lungentrübung manifestiert, können eine längere Intubation oder VA-ECMO-Unterstützung erforderlich machen. Eine Funktionsstörung des rechten Ventrikels kann neben anderen Komplikationen wie nosokomialen Pneumonien auch zum Einsatz von ECMO führen. Eine erneute Thrombose in endarteriektomierten Bereichen ist selten, und obwohl HCA die Wachsamkeit vorübergehend verringern kann, wurde sein Einsatz nicht mit anhaltenden kognitiven Dysfunktionen in Verbindung gebracht (
17
,
19
,
26
).Bei unbehandelter CTEPH liegt die 3-Jahres-Überlebensrate bei 70 %, wobei dies je nach hämodynamischer Schwere variiert (
17
,
27
). Durch PEA wird die Prognose signifikant verbessert, die 3-Jahres-Überlebensrate beträgt 89 % (
17
). Die chirurgische Sterblichkeitsrate hat sich drastisch von >20 % auf <5 % reduziert und ist nun mit anderen Herzoperationen vergleichbar (
26
,
28
,
29
). Nach PEA zeigen sich sofortige hämodynamische Verbesserungen, darunter ein verringerter pulmonalarterieller Druck (PAP), wobei innerhalb von drei bis zwölf Monaten eine Umgestaltung des rechten Ventrikels und eine Leistungssteigerung auftreten. Ein Jahr nach PEA sind eine signifikante Verringerung des pulmonalvaskulären Widerstands (PVR), Verbesserungen der 6-Minuten-Gehstrecke und Verbesserungen der NYHA-Funktionsklasse zu beobachten. 17–51 % der Patienten leiden an einer anhaltenden PH, häufig aufgrund von Mikrovaskularerkrankungen, technischen Herausforderungen bei der Entfernung distalen Materials oder Komorbiditäten. Dennoch erfahren diese Patienten im Allgemeinen eine klinische Besserung (
20
,
24
).2.2 Interventionelle Behandlung: Ballon-Pulmonalisangioplastie (BPA)Die pulmonale Ballonangioplastie (BPA) ist ein interventionelles Verfahren, das in mehreren Sitzungen durchgeführt wird und sich besonders zur Behandlung distaler subsegmentaler pulmonalarterieller Obstruktionen eignet, die sich einem chirurgischen Eingriff nicht unterziehen lassen. Kurzfristig hat die BPA im Vergleich zur Pharmakotherapie eine bessere Wirkung auf die Hämodynamik gezeigt, darunter eine Verbesserung der 6-Minuten-Gehstrecke, des pulmonalvaskulären Widerstands (PVR) und des mittleren pulmonalarteriellen Drucks (mPAP) (
15
,
30
). Daher ist die BPA typischerweise Patienten mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) vorbehalten, die keine Kandidaten für eine pulmonale Endarteriektomie (PEA) sind, oder die nach der Operation eine Rest-PH aufweisen (
31
). Die primäre Indikation für die BPA besteht in Szenarien, in denen PEA keine Option ist und sich Medikamente als unwirksam erwiesen haben. Für ihre Durchführung sind bestimmte Kriterien festgelegt (
Tabelle 4
). Tbelle 4
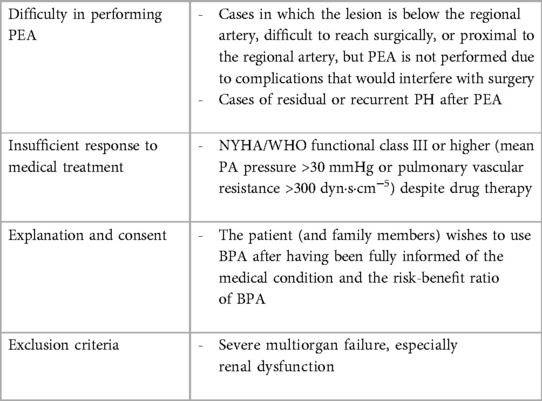 Tabelle 4. Indikationen für BPA. Das Potenzial einer sequentiellen Hybridstrategie, die PEA und BPA kombiniert, wurde hervorgehoben; Berichte deuten darauf hin, dass dieser Ansatz die Ergebnisse verbessern könnte (
32
). Darüber hinaus haben Studien keinen signifikanten Unterschied im Langzeitüberleben zwischen operierten und nicht operierten CTEPH-Patienten gezeigt. Dies lässt darauf schließen, dass ein kombinierter Ansatz aus PEA und BPA anstelle einer einzelnen Strategie zu signifikanten klinischen Verbesserungen der Prognose von CTEPH führen könnte (
33
). Obwohl weitere Forschung erforderlich ist, gibt es immer mehr Hinweise darauf, dass die Rolle von BPA über Patienten hinausgeht, bei denen PEA nicht in Frage kommt, und möglicherweise einer größeren Kohorte von CTEPH-Patienten in Verbindung mit PEA oder pharmakologischen Behandlungen zugutekommt (
34
). BPA hat sich bei der Linderung der Symptome sowie der Verbesserung der Lungenfunktion, der Belastungstoleranz und der hämodynamischen Parameter als erfolgreich erwiesen (
35
–
38
). Die spezifischen klinischen Merkmale, die eine Verbesserung der Belastungstoleranz nach BPA vorhersagen, müssen jedoch noch vollständig geklärt werden.Eine aktuelle Studie hat Licht auf Prädiktoren für eine verbesserte Belastungstoleranz nach BPA geworfen. Junges Alter, große Statur, kurze 6-Minuten-Gehstrecke (6MWD) vor dem Eingriff, hoher mPAP und hohe Lungenkapazität (%VC) wurden als Faktoren identifiziert, die mit besseren Belastungsergebnissen nach BPA in Zusammenhang stehen. Um die Auswirkungen von BPA auf die Belastungstoleranz genau vorhersagen zu können, ist eine umfassende präprozedurale Bewertung, die körperliche Aktivität, Atemfunktion und PH-Schweregrad umfasst, unerlässlich (
39
).Bei hämodynamisch und elektrisch stabilen Patienten ohne bestehende Infektionen kann eine Ballon-Pulmonalisangioplastie (BPA) nicht in Betracht gezogen werden, wenn echte absolute Kontraindikationen für eine Rechtsherzkatheterisierung (RHC) vorliegen – das heißt, wenn der Eingriff ein erhebliches Risiko für den Patienten darstellen würde, das nicht durch eine Optimierung vor dem Eingriff gemildert werden kann (wie etwa schwere Koagulopathie, mechanische Herzklappen, schwere Hypoxämie, unkooperative Patienten, Kontraindikationen für Sedierung oder Anästhesie oder andere schwere Komorbiditäten). Eine Vorbehandlung ist bei Patienten mit einer Allergie gegen Jodkontrastmittel erforderlich, einer relativen Kontraindikation. Faktoren wie aktive Infektionen, schwere chronisch obstruktive Lungenerkrankung, Hypoxämie, hämatologische Erkrankungen, die zu Blutungen oder Blutgerinnseln neigen, und unkontrollierte systemische Hypertonie müssen vor der Durchführung einer BPA beurteilt werden.Eine präoperative Optimierung ist entscheidend, um intra- und postoperative Risiken zu minimieren. Hämodynamische und systemische Zustände sollten stabilisiert werden. Die präoperative Gabe oraler Vasodilatatoren sollte fortgesetzt werden. Bei schweren Fällen mit niedrigem Herzzeitvolumen sollte eine inotrope Therapie mit Dobutamin erwogen werden (
40
,
41
).Die BPA wird unter Sedierung durchgeführt, wobei der femorale Zugang dem Zugang über die innere Jugularvene vorgezogen wird. Zielgefäße, die durch Perfusionsbildgebung, CT-Angiographie und intraprozedurale selektive Angiographie identifiziert wurden, werden über eine lange Schleuse durch eine kurze Schleuse erreicht, die in das betreffende Gefäßsegment eingeführt wird. Um einen umfassenden Nutzen zu erzielen, wird empfohlen, zuerst die Gefäße der Unterlappen zu behandeln, dann aber alle Lungensegmente abzubilden und zu behandeln. Die Antikoagulation während der BPA zielt auf eine aktivierte Gerinnungszeit von 200–250 s ab (
40
,
41
).Normalerweise sind 4–6 Sitzungen erforderlich, um alle Lungensegmente zu behandeln und eine Umgestaltung zu ermöglichen. Dabei geht es um die Beseitigung der PH, die Vorbeugung einer Rechtsherzinsuffizienz und die Verbesserung der Lebenserwartung. Das Erreichen eines mittleren pulmonalarteriellen Drucks von weniger als 25 mmHg und das Absetzen der häuslichen Sauerstofftherapie durch Verbesserung der Sauerstoffversorgung sind die primären Ziele (
40
,
41
).Die Komplikationsrate der BPA ist deutlich gesunken, was die Bedeutung der Durchführung dieses Verfahrens in erfahrenen Zentren unterstreicht. Komplikationen können durch Gefäßverletzungen aufgrund einer Drahtperforation, einer Ballondilatation und einer Kontrastmittelinjektion unter hohem Druck entstehen (
41
).In Studien, darunter einer in Japan und der RACE-Studie, wurde die Sicherheit und Wirksamkeit von BPA mit der von Riociguat bei inoperablen CTEPH-Patienten verglichen. Dabei zeigte sich, dass BPA trotz einer höheren Nebenwirkungsrate eine überlegene Verbesserung des mPAP und anderer hämodynamischer Parameter bewirkt (
42
,
43
).Es wurde auch gezeigt, dass BPA metabolische und systemische Störungen lindert, die mit Funktionsstörungen des rechten Ventrikels einhergehen. Signifikante Verbesserungen der körperlichen Leistungsfähigkeit, der Muskelkraft, der WHO-Funktionsklasse und möglicherweise der psychischen Lebensqualität nach der Rehabilitation nach BPA unterstreichen seine Vorteile (
44
–
47
).Zusammenfassend lässt sich sagen, dass BPA zwar eine Alternative für Patienten ist, die für PEA nicht in Frage kommen, es PEA jedoch nicht ersetzt, sondern eine mögliche Wahl für Kandidaten mit hohem Operationsrisiko darstellt. Mit fortschreitender Technik könnte BPA für bestimmte Patientengruppen bevorzugt werden, ergänzt durch die Aussicht auf Hybridverfahren, bei denen chirurgische und interventionelle Behandlungen für eine umfassende Versorgung kombiniert werden.
Tabelle 4. Indikationen für BPA. Das Potenzial einer sequentiellen Hybridstrategie, die PEA und BPA kombiniert, wurde hervorgehoben; Berichte deuten darauf hin, dass dieser Ansatz die Ergebnisse verbessern könnte (
32
). Darüber hinaus haben Studien keinen signifikanten Unterschied im Langzeitüberleben zwischen operierten und nicht operierten CTEPH-Patienten gezeigt. Dies lässt darauf schließen, dass ein kombinierter Ansatz aus PEA und BPA anstelle einer einzelnen Strategie zu signifikanten klinischen Verbesserungen der Prognose von CTEPH führen könnte (
33
). Obwohl weitere Forschung erforderlich ist, gibt es immer mehr Hinweise darauf, dass die Rolle von BPA über Patienten hinausgeht, bei denen PEA nicht in Frage kommt, und möglicherweise einer größeren Kohorte von CTEPH-Patienten in Verbindung mit PEA oder pharmakologischen Behandlungen zugutekommt (
34
). BPA hat sich bei der Linderung der Symptome sowie der Verbesserung der Lungenfunktion, der Belastungstoleranz und der hämodynamischen Parameter als erfolgreich erwiesen (
35
–
38
). Die spezifischen klinischen Merkmale, die eine Verbesserung der Belastungstoleranz nach BPA vorhersagen, müssen jedoch noch vollständig geklärt werden.Eine aktuelle Studie hat Licht auf Prädiktoren für eine verbesserte Belastungstoleranz nach BPA geworfen. Junges Alter, große Statur, kurze 6-Minuten-Gehstrecke (6MWD) vor dem Eingriff, hoher mPAP und hohe Lungenkapazität (%VC) wurden als Faktoren identifiziert, die mit besseren Belastungsergebnissen nach BPA in Zusammenhang stehen. Um die Auswirkungen von BPA auf die Belastungstoleranz genau vorhersagen zu können, ist eine umfassende präprozedurale Bewertung, die körperliche Aktivität, Atemfunktion und PH-Schweregrad umfasst, unerlässlich (
39
).Bei hämodynamisch und elektrisch stabilen Patienten ohne bestehende Infektionen kann eine Ballon-Pulmonalisangioplastie (BPA) nicht in Betracht gezogen werden, wenn echte absolute Kontraindikationen für eine Rechtsherzkatheterisierung (RHC) vorliegen – das heißt, wenn der Eingriff ein erhebliches Risiko für den Patienten darstellen würde, das nicht durch eine Optimierung vor dem Eingriff gemildert werden kann (wie etwa schwere Koagulopathie, mechanische Herzklappen, schwere Hypoxämie, unkooperative Patienten, Kontraindikationen für Sedierung oder Anästhesie oder andere schwere Komorbiditäten). Eine Vorbehandlung ist bei Patienten mit einer Allergie gegen Jodkontrastmittel erforderlich, einer relativen Kontraindikation. Faktoren wie aktive Infektionen, schwere chronisch obstruktive Lungenerkrankung, Hypoxämie, hämatologische Erkrankungen, die zu Blutungen oder Blutgerinnseln neigen, und unkontrollierte systemische Hypertonie müssen vor der Durchführung einer BPA beurteilt werden.Eine präoperative Optimierung ist entscheidend, um intra- und postoperative Risiken zu minimieren. Hämodynamische und systemische Zustände sollten stabilisiert werden. Die präoperative Gabe oraler Vasodilatatoren sollte fortgesetzt werden. Bei schweren Fällen mit niedrigem Herzzeitvolumen sollte eine inotrope Therapie mit Dobutamin erwogen werden (
40
,
41
).Die BPA wird unter Sedierung durchgeführt, wobei der femorale Zugang dem Zugang über die innere Jugularvene vorgezogen wird. Zielgefäße, die durch Perfusionsbildgebung, CT-Angiographie und intraprozedurale selektive Angiographie identifiziert wurden, werden über eine lange Schleuse durch eine kurze Schleuse erreicht, die in das betreffende Gefäßsegment eingeführt wird. Um einen umfassenden Nutzen zu erzielen, wird empfohlen, zuerst die Gefäße der Unterlappen zu behandeln, dann aber alle Lungensegmente abzubilden und zu behandeln. Die Antikoagulation während der BPA zielt auf eine aktivierte Gerinnungszeit von 200–250 s ab (
40
,
41
).Normalerweise sind 4–6 Sitzungen erforderlich, um alle Lungensegmente zu behandeln und eine Umgestaltung zu ermöglichen. Dabei geht es um die Beseitigung der PH, die Vorbeugung einer Rechtsherzinsuffizienz und die Verbesserung der Lebenserwartung. Das Erreichen eines mittleren pulmonalarteriellen Drucks von weniger als 25 mmHg und das Absetzen der häuslichen Sauerstofftherapie durch Verbesserung der Sauerstoffversorgung sind die primären Ziele (
40
,
41
).Die Komplikationsrate der BPA ist deutlich gesunken, was die Bedeutung der Durchführung dieses Verfahrens in erfahrenen Zentren unterstreicht. Komplikationen können durch Gefäßverletzungen aufgrund einer Drahtperforation, einer Ballondilatation und einer Kontrastmittelinjektion unter hohem Druck entstehen (
41
).In Studien, darunter einer in Japan und der RACE-Studie, wurde die Sicherheit und Wirksamkeit von BPA mit der von Riociguat bei inoperablen CTEPH-Patienten verglichen. Dabei zeigte sich, dass BPA trotz einer höheren Nebenwirkungsrate eine überlegene Verbesserung des mPAP und anderer hämodynamischer Parameter bewirkt (
42
,
43
).Es wurde auch gezeigt, dass BPA metabolische und systemische Störungen lindert, die mit Funktionsstörungen des rechten Ventrikels einhergehen. Signifikante Verbesserungen der körperlichen Leistungsfähigkeit, der Muskelkraft, der WHO-Funktionsklasse und möglicherweise der psychischen Lebensqualität nach der Rehabilitation nach BPA unterstreichen seine Vorteile (
44
–
47
).Zusammenfassend lässt sich sagen, dass BPA zwar eine Alternative für Patienten ist, die für PEA nicht in Frage kommen, es PEA jedoch nicht ersetzt, sondern eine mögliche Wahl für Kandidaten mit hohem Operationsrisiko darstellt. Mit fortschreitender Technik könnte BPA für bestimmte Patientengruppen bevorzugt werden, ergänzt durch die Aussicht auf Hybridverfahren, bei denen chirurgische und interventionelle Behandlungen für eine umfassende Versorgung kombiniert werden.
2.3 Medikamentöse TherapieDie medikamentöse Therapie spielt bei der Behandlung von chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) eine entscheidende Rolle, insbesondere bei der Behandlung der mikrovaskulären Erkrankung sowie bei Patienten, die als inoperabel gelten oder an anhaltender oder wiederkehrender PH nach pulmonaler Endarteriektomie (PEA) leiden. Die Grundlage der medikamentösen Therapie bei CTEPH umfasst eine lebenslange Antikoagulation zur Vorbeugung weiterer thromboembolischer Ereignisse sowie pulmonale Vasodilatatoren zur Behandlung der PH. Zur Behandlung einer Volumenüberlastung können Diuretika verschrieben werden, und bei Bedarf wird zusätzlicher Sauerstoff zur Korrektur der Hypoxämie eingesetzt.Drei bedeutende klinische Studien haben wesentlich zur Evidenzbasis für die pharmakologische Behandlung von CTEPH beigetragen: CHEST-1-Studie (Riociguat), CTREPH-Studie (Treprostinil) und MERIT-1-Studie (Macitentan), die später ausführlicher besprochen werden ( Tabelle 5 ). Tbelle 5
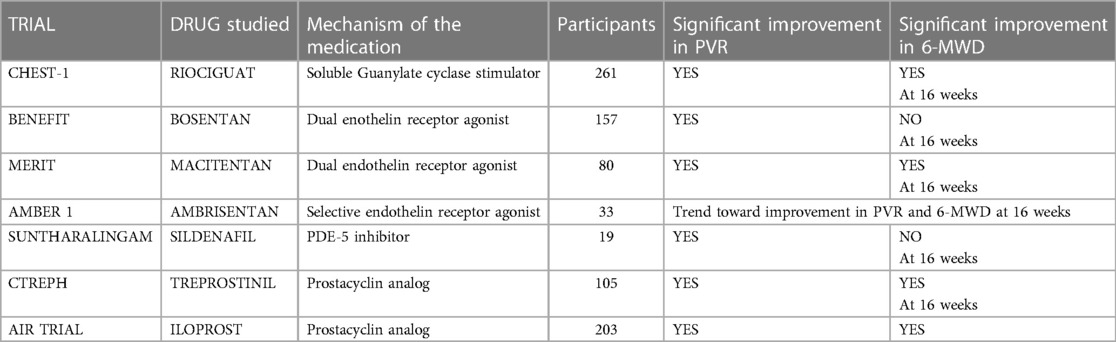 Tabelle 5. Wichtige Studien, in denen verschiedene Medikamente bei CTEPH untersucht wurden. Die medikamentöse Therapie von CTEPH besteht aus einer Antikoagulationstherapie und pulmonalen Vasodilatatoren, einschließlich Diuretika bei Volumenüberlastung und zusätzlicher Sauerstoffgabe bei Hypoxämie, falls angezeigt.2.3.1 AntikoagulationEine lebenslange Antikoagulationstherapie ist ein Eckpfeiler der Behandlung von chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) und ist auch nach einer pulmonalen Endarteriektomie (PEA) von entscheidender Bedeutung. Diese Empfehlung basiert auf der zugrunde liegenden Pathophysiologie der Krankheit, die durch wiederkehrende pulmonale Thromboembolien und unzureichende Gerinnselauflösung gekennzeichnet ist. Während die optimale Wahl des Antikoagulans aufgrund des Fehlens randomisierter kontrollierter Studien (RCTs) speziell für CTEPH noch nicht definiert ist, hat sich Warfarin als das von Experten empfohlene und am häufigsten eingesetzte Mittel herausgestellt. Es wird empfohlen, Warfarin auf unbestimmte Zeit zu verabreichen und dabei ein Prothrombinzeit-International Normalized Ratio (PT-INR) zwischen 2,0 und 3,0 aufrechtzuerhalten (
48
).In den letzten Jahren hat die Verwendung direkter oraler Antikoagulanzien (DOACs) als Alternative zu Warfarin zugenommen, obwohl ihre Wirksamkeit bei CTEPH nicht eindeutig belegt ist. Eine retrospektive Fallserie aus Großbritannien und das multizentrische prospektive Register (EXPERT) zeigten bei CTEPH-Patienten ähnliche Blutungsraten bei Warfarin und DOACs. Bei Patienten, die neuartige orale Antikoagulanzien (NOACs) einnahmen, traten jedoch häufiger wiederkehrende venöse Thromboembolien auf (
49
).Bei Patienten mit CTEPH und gleichzeitigem Antiphospholipid-Syndrom – eine Erkrankung, die etwa 10 % der CTEPH-Patienten betrifft – gilt die Therapie mit Vitamin-K-Antagonisten (VKA) als sicherer als DOACs wie Rivaroxaban (
50
). Daher werden in diesen Fällen VKAs zur Antikoagulation empfohlen (
51
).Die jüngsten Fortschritte in der Antikoagulationstherapie bei chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) werden in zwei bedeutenden Studien hervorgehoben. Die AXADIA-AFNET 8-Studie deutet darauf hin, dass Apixaban für Patienten mit Vorhofflimmern, die sich einer chronischen Hämodialyse unterziehen, eine sichere und effektive Alternative zu Vitamin-K-Antagonisten darstellt, und weist auch auf potenzielle Vorteile im Zusammenhang mit CTEPH hin (
52
). Die Studie könnte wertvolle Daten zur Risikostratifizierung bei Patienten liefern, die eine Antikoagulation benötigen. Dies kann bei der Entwicklung von Strategien zur Identifizierung der CTEPH-Patienten helfen, die am meisten von bestimmten Antikoagulanzien profitieren könnten. Darüber hinaus kann das Verständnis, wie unterschiedliche Patientenmerkmale die Wirksamkeit und Sicherheit der Antikoagulationstherapie bei Vorhofflimmern (AF) beeinflussen, dazu beitragen, ähnliche Überlegungen bei CTEPH-Patienten anzustellen, insbesondere da beide Populationen überlappende Komorbiditäten wie Bluthochdruck und Herzerkrankungen aufweisen können. Abschließend sollte betont werden, dass die Ergebnisse hinsichtlich Schlaganfall-, systemischer Embolie- und Blutungsrisiken bei Patienten mit Vorhofflimmern eine vergleichende Perspektive darauf bieten können, was in einer Patientengruppe zu erwarten ist, die eine langfristige Antikoagulation benötigt, wie z. B. bei CTEPH. Unterdessen kam die KABUKI-Studie, eine multizentrische, einfach verblindete randomisierte Studie, zu dem Ergebnis, dass Edoxaban bei der Vorbeugung einer Verschlechterung des pulmonalvaskulären Widerstands bei CTEPH-Patienten Warfarin nicht unterlegen ist und ein ähnliches Sicherheitsprofil aufweist (
53
). Diese Erkenntnisse tragen zur sich weiterentwickelnden Landschaft der Antikoagulationstherapie bei CTEPH bei und legen nahe, dass direkte orale Antikoagulanzien wie Apixaban und Edoxaban brauchbare Alternativen zur traditionellen Therapie darstellen könnten, was weitere Untersuchungen rechtfertigt.2.3.2 ProstacyclinProstacyclin, ein potenter Vasodilatator und Inhibitor der Thrombozytenaggregation sowie der Proliferation glatter Muskelzellen, der von Endothelzellen gebildet wird, spielt eine bedeutende Rolle in der Physiologie der Lungengefäße. Bei Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) sind seine Konzentrationen verringert, was zur Anwendung von Prostanoid-Medikamenten bei der Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) führt (
54
).In der CTREPH-Studie wurde die Wirksamkeit von Treprostinil, einem Prostazyklin-Analogon, bei CTEPH-Patienten untersucht, die entweder nicht für eine pulmonale Endarteriektomie (PEA) in Frage kamen oder sich gegen den chirurgischen Eingriff entschieden. In dieser Studie wurden die Wirkungen einer niedrig dosierten (3 ng/kg/min) vs. einer hoch dosierten (30 ng/kg/min) subkutanen Treprostinil-Infusion über einen Zeitraum von 24 Wochen verglichen. Das primäre Ergebnis, eine Veränderung der 6-Minuten-Gehstrecke (6-MWD), zeigte bei der höheren Treprostinil-Dosis eine signifikante Verbesserung von +40 m, was auf den günstigen Einfluss von Treprostinil auf die körperliche Leistungsfähigkeit hindeutet (
55
). Folglich wurde Treprostinil in der Europäischen Union zur Behandlung von CTEPH bei Patienten der WHO-Funktionsklasse III–IV zugelassen, die entweder an einer inoperablen Erkrankung oder an anhaltender/rezidivierender PH nach PEA leiden und die Empfehlung IIb B erhalten (
2
).Darüber hinaus hat eine langfristige intravenöse Epoprostenoltherapie bei inoperablen CTEPH-Patienten vielversprechende Ergebnisse gezeigt. Eine Studie mit 27 Patienten ergab über einen Beobachtungszeitraum von 20 Monaten deutliche Verbesserungen der körperlichen Belastbarkeit und des Herzindex (CI). Nach einer dreimonatigen Behandlung kam es bei 11 von 23 Patienten zu einer bemerkenswerten Verbesserung der NYHA-Funktionsklasse, einer Zunahme der 6MWD um 66 m und signifikanten hämodynamischen Verbesserungen [mittlerer pulmonalarterieller Druck (mPAP), Herzindex und totaler pulmonaler Widerstand], was alles auf die Wirksamkeit der Therapie hinweist. Die Überlebensraten nach 1, 2 und 3 Jahren lagen bei 73 %, 59 % bzw. 41 %, was den potenziellen lebensverlängernden Nutzen von Epoprostenol bei dieser Patientengruppe unterstreicht (
56
).2.3.3 SelexipagSelexipag, ein oraler selektiver Agonist für den Prostacyclin-Rezeptor (IP-Rezeptor) mit einer nicht-prostanoiden Struktur, und sein Metabolit MRE-269 weisen eine hohe Selektivität für den IP-Rezeptor auf und steigern die Produktion von zyklischem Adenosinmonophosphat (cAMP). Dieser Mechanismus fördert die Entspannung der Gefäßglattmuskulatur (
57
,
58
). Frühere Studien an Patienten mit pulmonaler arterieller Hypertonie (PAH) haben die Wirksamkeit von Selexipag bei der Verringerung des Morbiditäts-/Mortalitätsrisikos gezeigt, was zu seiner Zulassung zur PAH-Behandlung in verschiedenen Regionen führte, darunter den Vereinigten Staaten, der Europäischen Union und Japan (
59
).Im Zusammenhang mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) wurde in einer multizentrischen, doppelblinden, placebokontrollierten Phase-3-Studie in Japan die Wirksamkeit und Sicherheit von Selexipag bei Patienten mit inoperabler CTEPH oder anhaltender/rezidivierender PH nach PEA und/oder BPA untersucht (
60
). Die Behandlung begann mit 200 μg Selexipag zweimal täglich und wurde je nach Verträglichkeit auf 1.600 μg zweimal täglich gesteigert. Die Dosis wurde alle drei Tage in 200-μg-Schritten auf insgesamt sechs Dosen über 20 Wochen angepasst, wobei die maximal verträgliche Dosis während der letzten 8 Wochen beibehalten wurde.In der Studie wurde vor allem der Einfluss auf den pulmonalvaskulären Widerstand (PVR) gemessen, einen kritischen hämodynamischen Parameter der PH, der mit der Langzeitprognose korreliert. Die Senkung des PVR durch Selexipag steht im Einklang mit Ergebnissen aus früheren Studien zu pulmonalvasodilatatoren bei CTEPH (
12
,
61
,
62
) und unterstützt Hinweise aus einer früheren Proof-of-Concept-Studie, die auf das Potenzial von Selexipag zur Verbesserung der Hämodynamik bei japanischen CTEPH-Patienten hindeutet (
63
). Neben dem PVR wurden Verbesserungen bei anderen hämodynamischen Parametern festgestellt, darunter PVR-Index, Herzindex, totaler pulmonaler Widerstand und gemischtvenöse Sauerstoffsättigung (SvO2).Trotz dieser vielversprechenden Ergebnisse zeigten sich bei sekundären Endpunkten wie der 6-Minuten-Gehstrecke (6MWD) und der WHO-Funktionsklasse keine signifikanten Unterschiede zwischen der Selexipag- und der Placebo-Gruppe, was möglicherweise an der Stichprobengröße der Studie liegt.Die Studie kommt zu dem Schluss, dass Selexipag sowohl gut verträglich als auch sicher ist und hämodynamische Vorteile für CTEPH-Patienten bietet, für die PEA nicht infrage kommt, oder für Patienten mit anhaltender PH nach PEA und/oder BPA. Allerdings wurde keine Verbesserung der körperlichen Leistungsfähigkeit beobachtet. Diese Ergebnisse unterstreichen die Notwendigkeit weiterer groß angelegter Forschung, um die Rolle von Selexipag bei der Behandlung von CTEPH endgültig zu etablieren.2.3.4 Endothelin-Rezeptor-AntagonistenEndothelin-1, ein potenter Vasokonstriktor und Mitogen für glatte Muskelzellen, der von Endothelzellen produziert wird, wurde in erhöhten Plasmaspiegeln bei Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) und chronischer thromboembolischer pulmonaler Hypertonie (CTEPH) nachgewiesen (
64
). Endothelin-Rezeptor-Antagonisten (ERAs) mildern die Effekte von Endothelin-1, indem sie entweder selektiv Typ-A-Rezeptoren oder nicht-selektiv sowohl Typ-A- als auch Typ-B-Rezeptoren blockieren und so die vasokonstriktiven und mitogenen Reaktionen hemmen (
65
,
66
).Die BENEFIT-Studie, in der der duale Endothelin-Rezeptor-Inhibitor Bosentan bei CTEPH-Patienten untersucht wurde, kam zu gemischten Ergebnissen. Während Bosentan den pulmonalvaskulären Widerstand (PVR) effektiv reduzierte, hatte es über 16 Behandlungswochen bei 77 Patienten mit symptomatischer CTEPH, die entweder inoperabel waren oder länger als 6 Monate nach PEA an anhaltender PH litten, keinen signifikanten Einfluss auf die 6-Minuten-Gehstrecke (6MWD). Die Studie ergab auch keinen statistisch signifikanten Unterschied in der Zeit bis zur klinischen Verschlechterung zwischen mit Bosentan behandelten Patienten und der Placebogruppe (80 Patienten) (
11
).Reesnik et al. beobachteten verbesserte klinische und hämodynamische Ergebnisse durch eine Bosentan-Behandlung vor einer Endarteriektomie. Die begrenzte Anzahl der bislang durchgeführten Studien kann den Nutzen dieser präoperativen medizinischen Intervention jedoch nicht schlüssig belegen (
67
).Macitentan, das als Verbesserung gegenüber Bosentan mit dualem ETRA/ETRB-Antagonismus entwickelt wurde, wurde zur Behandlung von PAH zugelassen. In der Phase-2-Studie MERIT-1, einer doppelblinden, randomisierten, placebokontrollierten Studie, wurde die Wirksamkeit von 10 mg Macitentan bei 80 Patienten mit inoperabler CTEPH untersucht. Nach 16 Wochen war der geometrische Mittelwert des PVR in der Macitentan-Gruppe auf 73,0 % des Ausgangswerts gesunken, im Vergleich zu 87,2 % in der Placebogruppe, was auf eine signifikante Verbesserung des PVR bei Patienten mit inoperabler CTEPH hindeutet (
13
). Darüber hinaus ist es sicherlich wichtig hervorzuheben, dass 61 % der in die MERIT-Studie aufgenommenen Patienten einen PDE5i einnahmen, da dies bedeutet, dass die Kombination von Macitentan mit einem PDE5i bei inoperabler CTEPH von Nutzen sein kann.Derzeit läuft eine randomisierte kontrollierte Studie der Phase 3 zur Beurteilung der Sicherheit und Wirksamkeit von 75 mg Macitentan bei Patienten mit inoperabler oder anhaltender/rezidivierender CTEPH (NCT04271475). Diese Studie verspricht weitere Erkenntnisse zum therapeutischen Potenzial von Macitentan bei der Behandlung von CTEPH.2.3.5 Phosphodiesterase-Typ-5-InhibitorenStickstoffmonoxid (NO) ist ein wichtiger endogener Vasodilatator, der von Endothelzellen produziert wird und eine zentrale Rolle bei der Hemmung der Thrombozytenaggregation und der Proliferation glatter Muskelzellen spielt. NO aktiviert die lösliche Guanylatcyclase (sGC) zur Synthese von zyklischem Guanosinmonophosphat (cGMP), was zur Entspannung der glatten Muskulatur führt (
12
). Phosphodiesterase-5 (PDE5)-Hemmer wie Sildenafil verstärken die Vasodilatation, indem sie den Abbau von cGMP verhindern. Ihre Wirksamkeit hängt jedoch von der Anwesenheit von NO ab, was eine begrenzte Wirksamkeit in Lungenregionen mit vermindertem NO-Spiegel bedeutet (
68
,
69
).Sildenafil wurde insbesondere auf seine Wirkung auf chronisch thromboembolische pulmonale Hypertonie (CTEPH) untersucht. In einer offenen, unkontrollierten Studie von Reichenberger et al. wurden 104 Patienten mit inoperabler CTEPH dreimal täglich mit Sildenafil in einer Dosierung von 50 mg behandelt. Die Ergebnisse zeigten eine signifikante Reduktion des pulmonalvaskulären Widerstandes (PVR) von einem Ausgangswert von 863 ± 38 dyn·s·cm −5 auf 759 ± 62 dyn·s·cm −5 nach drei Monaten ( p = 0,0002 vs. Ausgangswert), zusammen mit einer bemerkenswerten Erhöhung der 6-Minuten-Gehstrecke (6MWD) von 310 ± 11 m zu Beginn auf 361 ± 15 m nach drei Monaten ( p = 0,0001 vs. Ausgangswert) und 366 ± 18 m nach zwölf Monaten ( p = 0,0005 vs. Ausgangswert), was auf wesentliche hämodynamische und funktionelle Verbesserungen hindeutet (
70
).Während Sildenafil bei inoperabler CTEPH aufgrund des Fehlens randomisierter kontrollierter Studien (RCTs) oder Registerdaten, die seine Wirksamkeit belegen, außerhalb der Zulassung eingesetzt wird, hat sich die orale Kombinationstherapie mit PDE5-Hemmern und Endothelin-Rezeptorantagonisten als gängige Methode zur Behandlung von Patienten mit CTEPH mit schweren hämodynamischen Beeinträchtigungen etabliert (
71
). Diese Praxis unterstreicht die Integration verschiedener pharmakologischer Strategien zur Linderung der Symptome und Verbesserung der Lebensqualität der Patienten mit dieser herausfordernden Erkrankung.2.3.6 Löslicher Guanylatcyclase-StimulatorObwohl in zahlreichen randomisierten kontrollierten Studien die Wirksamkeit und Sicherheit verschiedener Behandlungen untersucht wurde, ist Riociguat das einzige Medikament, das von der FDA speziell sowohl für pulmonale Hypertonie (PH) als auch für chronisch thromboembolische pulmonale Hypertonie (CTEPH) zugelassen wurde. Riociguat, ein Stimulator der löslichen Guanylatcyclase (sGC), wird zur Behandlung von inoperabler CTEPH sowie persistierender/rezidivierender CTEPH nach pulmonaler Endarteriektomie (PEA) empfohlen und erhält IB-Empfehlungen (
2
,
72
). Dieses Medikament wirkt, indem es sGC direkt stimuliert, die cGMP-Werte unabhängig von Stickstoffmonoxid (NO) erhöht und die Empfindlichkeit der Gefäßwandzellen gegenüber endogenem NO steigert und so die Vasodilatation in schlecht belüfteten Lungenbereichen erleichtert (
73
).Erste Berichte von Ghofrani et al. aus dem Jahr 2010 zeigten, dass Riociguat die körperliche Leistungsfähigkeit bei Patienten mit CTEPH und PAH über einen Zeitraum von 12 Wochen verbessern und die Krankheitssymptome reduzieren konnte (
74
). Die PATENT-1-Studie bestätigte den Nutzen von Riociguat bei PAH-Patienten weiter und zeigte signifikante Verbesserungen der körperlichen Leistungsfähigkeit, der WHO-Funktionsklasse, des pulmonal-vaskulären Widerstands (PVR) und der NT-proBNP-Werte (
75
). Die langfristige Sicherheit und Wirksamkeit wurden in der PATENT-2-Studie bestätigt, in der es über ein Jahr hinweg zu anhaltenden Verbesserungen der funktionellen und körperlichen Leistungsfähigkeit kam (
76
).In der PATENT PLUS-Studie wurden die additiven Effekte von Riociguat und PDE5-Hemmern bei PAH-Patienten untersucht, die mit Sildenafil behandelt wurden. Zwischen den beiden Gruppen konnten keine signifikanten Unterschiede hinsichtlich der hämodynamischen Verbesserungen oder der körperlichen Leistungsfähigkeit festgestellt werden (
77
). In der CHEST-1-Studie an inoperablen CTEPH-Patienten wurden unter Riociguat-Behandlung ähnliche Verbesserungen der körperlichen Leistungsfähigkeit, der WHO-Funktionsklasse, des PVR und der NT-proBNP-Werte berichtet (
12
). Der langfristige Nutzen wurde in der CHEST-2-Studie bestätigt (
78
).Trotz dieser positiven Ergebnisse wurden in den Studien jedoch keine Parameter der Rechtsherzfunktionsstörung direkt ausgewertet, die für die Vorhersage des Überlebens entscheidend sind. Eine umfassende Analyse von Marra A. et al. mit Patienten aus den Studien PATENT-1, PATENT PLUS, EAS und CHEST zeigte im Verlauf einer 12-monatigen Riociguat-Therapie signifikante Reduktionen der rechtsventrikulären Dimensionen und Verbesserungen der TAPSE (
79
).Es ist wichtig zu beachten, dass es keine direkten Vergleichsstudien zwischen Riociguat und anderen medizinischen Therapien für CTEPH gibt, da Unterschiede im Studiendesign direkte Vergleiche erschweren. Die Studien CHEST-1, CTREPH und MERIT-1 hatten jeweils unterschiedliche Patientenpopulationen und Basistherapiezuteilungen, was zur Variabilität der Patientendemographie und der Schwere der Erkrankung beitrug (
6
,
12
,
13
,
54
).Riociguat ist die einzige von der FDA zugelassene Behandlung für inoperable CTEPH oder post-PEA-Rest-PH (
72
). Während eine orale Kombinationstherapie, einschließlich PDE5-Hemmern und Endothelin-Rezeptor-Antagonisten, bei CTEPH-Patienten mit schweren hämodynamischen Störungen weit verbreitet ist (
71
), bleibt die Rolle von Vasodilatatoren als präoperative Behandlung umstritten, da sie den chirurgischen Eingriff potenziell verzögern kann (
67
,
80
). Im Gegensatz dazu kann bei Patienten, bei denen eine BPA in Betracht gezogen wird, eine medikamentöse Vorbehandlung die Lungenhämodynamik und die Verfahrenssicherheit verbessern, obwohl diese Empfehlung auf Evidenz von sehr geringer Qualität beruht (
2
).
Tabelle 5. Wichtige Studien, in denen verschiedene Medikamente bei CTEPH untersucht wurden. Die medikamentöse Therapie von CTEPH besteht aus einer Antikoagulationstherapie und pulmonalen Vasodilatatoren, einschließlich Diuretika bei Volumenüberlastung und zusätzlicher Sauerstoffgabe bei Hypoxämie, falls angezeigt.2.3.1 AntikoagulationEine lebenslange Antikoagulationstherapie ist ein Eckpfeiler der Behandlung von chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) und ist auch nach einer pulmonalen Endarteriektomie (PEA) von entscheidender Bedeutung. Diese Empfehlung basiert auf der zugrunde liegenden Pathophysiologie der Krankheit, die durch wiederkehrende pulmonale Thromboembolien und unzureichende Gerinnselauflösung gekennzeichnet ist. Während die optimale Wahl des Antikoagulans aufgrund des Fehlens randomisierter kontrollierter Studien (RCTs) speziell für CTEPH noch nicht definiert ist, hat sich Warfarin als das von Experten empfohlene und am häufigsten eingesetzte Mittel herausgestellt. Es wird empfohlen, Warfarin auf unbestimmte Zeit zu verabreichen und dabei ein Prothrombinzeit-International Normalized Ratio (PT-INR) zwischen 2,0 und 3,0 aufrechtzuerhalten (
48
).In den letzten Jahren hat die Verwendung direkter oraler Antikoagulanzien (DOACs) als Alternative zu Warfarin zugenommen, obwohl ihre Wirksamkeit bei CTEPH nicht eindeutig belegt ist. Eine retrospektive Fallserie aus Großbritannien und das multizentrische prospektive Register (EXPERT) zeigten bei CTEPH-Patienten ähnliche Blutungsraten bei Warfarin und DOACs. Bei Patienten, die neuartige orale Antikoagulanzien (NOACs) einnahmen, traten jedoch häufiger wiederkehrende venöse Thromboembolien auf (
49
).Bei Patienten mit CTEPH und gleichzeitigem Antiphospholipid-Syndrom – eine Erkrankung, die etwa 10 % der CTEPH-Patienten betrifft – gilt die Therapie mit Vitamin-K-Antagonisten (VKA) als sicherer als DOACs wie Rivaroxaban (
50
). Daher werden in diesen Fällen VKAs zur Antikoagulation empfohlen (
51
).Die jüngsten Fortschritte in der Antikoagulationstherapie bei chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) werden in zwei bedeutenden Studien hervorgehoben. Die AXADIA-AFNET 8-Studie deutet darauf hin, dass Apixaban für Patienten mit Vorhofflimmern, die sich einer chronischen Hämodialyse unterziehen, eine sichere und effektive Alternative zu Vitamin-K-Antagonisten darstellt, und weist auch auf potenzielle Vorteile im Zusammenhang mit CTEPH hin (
52
). Die Studie könnte wertvolle Daten zur Risikostratifizierung bei Patienten liefern, die eine Antikoagulation benötigen. Dies kann bei der Entwicklung von Strategien zur Identifizierung der CTEPH-Patienten helfen, die am meisten von bestimmten Antikoagulanzien profitieren könnten. Darüber hinaus kann das Verständnis, wie unterschiedliche Patientenmerkmale die Wirksamkeit und Sicherheit der Antikoagulationstherapie bei Vorhofflimmern (AF) beeinflussen, dazu beitragen, ähnliche Überlegungen bei CTEPH-Patienten anzustellen, insbesondere da beide Populationen überlappende Komorbiditäten wie Bluthochdruck und Herzerkrankungen aufweisen können. Abschließend sollte betont werden, dass die Ergebnisse hinsichtlich Schlaganfall-, systemischer Embolie- und Blutungsrisiken bei Patienten mit Vorhofflimmern eine vergleichende Perspektive darauf bieten können, was in einer Patientengruppe zu erwarten ist, die eine langfristige Antikoagulation benötigt, wie z. B. bei CTEPH. Unterdessen kam die KABUKI-Studie, eine multizentrische, einfach verblindete randomisierte Studie, zu dem Ergebnis, dass Edoxaban bei der Vorbeugung einer Verschlechterung des pulmonalvaskulären Widerstands bei CTEPH-Patienten Warfarin nicht unterlegen ist und ein ähnliches Sicherheitsprofil aufweist (
53
). Diese Erkenntnisse tragen zur sich weiterentwickelnden Landschaft der Antikoagulationstherapie bei CTEPH bei und legen nahe, dass direkte orale Antikoagulanzien wie Apixaban und Edoxaban brauchbare Alternativen zur traditionellen Therapie darstellen könnten, was weitere Untersuchungen rechtfertigt.2.3.2 ProstacyclinProstacyclin, ein potenter Vasodilatator und Inhibitor der Thrombozytenaggregation sowie der Proliferation glatter Muskelzellen, der von Endothelzellen gebildet wird, spielt eine bedeutende Rolle in der Physiologie der Lungengefäße. Bei Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) sind seine Konzentrationen verringert, was zur Anwendung von Prostanoid-Medikamenten bei der Behandlung der chronisch thromboembolischen pulmonalen Hypertonie (CTEPH) führt (
54
).In der CTREPH-Studie wurde die Wirksamkeit von Treprostinil, einem Prostazyklin-Analogon, bei CTEPH-Patienten untersucht, die entweder nicht für eine pulmonale Endarteriektomie (PEA) in Frage kamen oder sich gegen den chirurgischen Eingriff entschieden. In dieser Studie wurden die Wirkungen einer niedrig dosierten (3 ng/kg/min) vs. einer hoch dosierten (30 ng/kg/min) subkutanen Treprostinil-Infusion über einen Zeitraum von 24 Wochen verglichen. Das primäre Ergebnis, eine Veränderung der 6-Minuten-Gehstrecke (6-MWD), zeigte bei der höheren Treprostinil-Dosis eine signifikante Verbesserung von +40 m, was auf den günstigen Einfluss von Treprostinil auf die körperliche Leistungsfähigkeit hindeutet (
55
). Folglich wurde Treprostinil in der Europäischen Union zur Behandlung von CTEPH bei Patienten der WHO-Funktionsklasse III–IV zugelassen, die entweder an einer inoperablen Erkrankung oder an anhaltender/rezidivierender PH nach PEA leiden und die Empfehlung IIb B erhalten (
2
).Darüber hinaus hat eine langfristige intravenöse Epoprostenoltherapie bei inoperablen CTEPH-Patienten vielversprechende Ergebnisse gezeigt. Eine Studie mit 27 Patienten ergab über einen Beobachtungszeitraum von 20 Monaten deutliche Verbesserungen der körperlichen Belastbarkeit und des Herzindex (CI). Nach einer dreimonatigen Behandlung kam es bei 11 von 23 Patienten zu einer bemerkenswerten Verbesserung der NYHA-Funktionsklasse, einer Zunahme der 6MWD um 66 m und signifikanten hämodynamischen Verbesserungen [mittlerer pulmonalarterieller Druck (mPAP), Herzindex und totaler pulmonaler Widerstand], was alles auf die Wirksamkeit der Therapie hinweist. Die Überlebensraten nach 1, 2 und 3 Jahren lagen bei 73 %, 59 % bzw. 41 %, was den potenziellen lebensverlängernden Nutzen von Epoprostenol bei dieser Patientengruppe unterstreicht (
56
).2.3.3 SelexipagSelexipag, ein oraler selektiver Agonist für den Prostacyclin-Rezeptor (IP-Rezeptor) mit einer nicht-prostanoiden Struktur, und sein Metabolit MRE-269 weisen eine hohe Selektivität für den IP-Rezeptor auf und steigern die Produktion von zyklischem Adenosinmonophosphat (cAMP). Dieser Mechanismus fördert die Entspannung der Gefäßglattmuskulatur (
57
,
58
). Frühere Studien an Patienten mit pulmonaler arterieller Hypertonie (PAH) haben die Wirksamkeit von Selexipag bei der Verringerung des Morbiditäts-/Mortalitätsrisikos gezeigt, was zu seiner Zulassung zur PAH-Behandlung in verschiedenen Regionen führte, darunter den Vereinigten Staaten, der Europäischen Union und Japan (
59
).Im Zusammenhang mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) wurde in einer multizentrischen, doppelblinden, placebokontrollierten Phase-3-Studie in Japan die Wirksamkeit und Sicherheit von Selexipag bei Patienten mit inoperabler CTEPH oder anhaltender/rezidivierender PH nach PEA und/oder BPA untersucht (
60
). Die Behandlung begann mit 200 μg Selexipag zweimal täglich und wurde je nach Verträglichkeit auf 1.600 μg zweimal täglich gesteigert. Die Dosis wurde alle drei Tage in 200-μg-Schritten auf insgesamt sechs Dosen über 20 Wochen angepasst, wobei die maximal verträgliche Dosis während der letzten 8 Wochen beibehalten wurde.In der Studie wurde vor allem der Einfluss auf den pulmonalvaskulären Widerstand (PVR) gemessen, einen kritischen hämodynamischen Parameter der PH, der mit der Langzeitprognose korreliert. Die Senkung des PVR durch Selexipag steht im Einklang mit Ergebnissen aus früheren Studien zu pulmonalvasodilatatoren bei CTEPH (
12
,
61
,
62
) und unterstützt Hinweise aus einer früheren Proof-of-Concept-Studie, die auf das Potenzial von Selexipag zur Verbesserung der Hämodynamik bei japanischen CTEPH-Patienten hindeutet (
63
). Neben dem PVR wurden Verbesserungen bei anderen hämodynamischen Parametern festgestellt, darunter PVR-Index, Herzindex, totaler pulmonaler Widerstand und gemischtvenöse Sauerstoffsättigung (SvO2).Trotz dieser vielversprechenden Ergebnisse zeigten sich bei sekundären Endpunkten wie der 6-Minuten-Gehstrecke (6MWD) und der WHO-Funktionsklasse keine signifikanten Unterschiede zwischen der Selexipag- und der Placebo-Gruppe, was möglicherweise an der Stichprobengröße der Studie liegt.Die Studie kommt zu dem Schluss, dass Selexipag sowohl gut verträglich als auch sicher ist und hämodynamische Vorteile für CTEPH-Patienten bietet, für die PEA nicht infrage kommt, oder für Patienten mit anhaltender PH nach PEA und/oder BPA. Allerdings wurde keine Verbesserung der körperlichen Leistungsfähigkeit beobachtet. Diese Ergebnisse unterstreichen die Notwendigkeit weiterer groß angelegter Forschung, um die Rolle von Selexipag bei der Behandlung von CTEPH endgültig zu etablieren.2.3.4 Endothelin-Rezeptor-AntagonistenEndothelin-1, ein potenter Vasokonstriktor und Mitogen für glatte Muskelzellen, der von Endothelzellen produziert wird, wurde in erhöhten Plasmaspiegeln bei Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) und chronischer thromboembolischer pulmonaler Hypertonie (CTEPH) nachgewiesen (
64
). Endothelin-Rezeptor-Antagonisten (ERAs) mildern die Effekte von Endothelin-1, indem sie entweder selektiv Typ-A-Rezeptoren oder nicht-selektiv sowohl Typ-A- als auch Typ-B-Rezeptoren blockieren und so die vasokonstriktiven und mitogenen Reaktionen hemmen (
65
,
66
).Die BENEFIT-Studie, in der der duale Endothelin-Rezeptor-Inhibitor Bosentan bei CTEPH-Patienten untersucht wurde, kam zu gemischten Ergebnissen. Während Bosentan den pulmonalvaskulären Widerstand (PVR) effektiv reduzierte, hatte es über 16 Behandlungswochen bei 77 Patienten mit symptomatischer CTEPH, die entweder inoperabel waren oder länger als 6 Monate nach PEA an anhaltender PH litten, keinen signifikanten Einfluss auf die 6-Minuten-Gehstrecke (6MWD). Die Studie ergab auch keinen statistisch signifikanten Unterschied in der Zeit bis zur klinischen Verschlechterung zwischen mit Bosentan behandelten Patienten und der Placebogruppe (80 Patienten) (
11
).Reesnik et al. beobachteten verbesserte klinische und hämodynamische Ergebnisse durch eine Bosentan-Behandlung vor einer Endarteriektomie. Die begrenzte Anzahl der bislang durchgeführten Studien kann den Nutzen dieser präoperativen medizinischen Intervention jedoch nicht schlüssig belegen (
67
).Macitentan, das als Verbesserung gegenüber Bosentan mit dualem ETRA/ETRB-Antagonismus entwickelt wurde, wurde zur Behandlung von PAH zugelassen. In der Phase-2-Studie MERIT-1, einer doppelblinden, randomisierten, placebokontrollierten Studie, wurde die Wirksamkeit von 10 mg Macitentan bei 80 Patienten mit inoperabler CTEPH untersucht. Nach 16 Wochen war der geometrische Mittelwert des PVR in der Macitentan-Gruppe auf 73,0 % des Ausgangswerts gesunken, im Vergleich zu 87,2 % in der Placebogruppe, was auf eine signifikante Verbesserung des PVR bei Patienten mit inoperabler CTEPH hindeutet (
13
). Darüber hinaus ist es sicherlich wichtig hervorzuheben, dass 61 % der in die MERIT-Studie aufgenommenen Patienten einen PDE5i einnahmen, da dies bedeutet, dass die Kombination von Macitentan mit einem PDE5i bei inoperabler CTEPH von Nutzen sein kann.Derzeit läuft eine randomisierte kontrollierte Studie der Phase 3 zur Beurteilung der Sicherheit und Wirksamkeit von 75 mg Macitentan bei Patienten mit inoperabler oder anhaltender/rezidivierender CTEPH (NCT04271475). Diese Studie verspricht weitere Erkenntnisse zum therapeutischen Potenzial von Macitentan bei der Behandlung von CTEPH.2.3.5 Phosphodiesterase-Typ-5-InhibitorenStickstoffmonoxid (NO) ist ein wichtiger endogener Vasodilatator, der von Endothelzellen produziert wird und eine zentrale Rolle bei der Hemmung der Thrombozytenaggregation und der Proliferation glatter Muskelzellen spielt. NO aktiviert die lösliche Guanylatcyclase (sGC) zur Synthese von zyklischem Guanosinmonophosphat (cGMP), was zur Entspannung der glatten Muskulatur führt (
12
). Phosphodiesterase-5 (PDE5)-Hemmer wie Sildenafil verstärken die Vasodilatation, indem sie den Abbau von cGMP verhindern. Ihre Wirksamkeit hängt jedoch von der Anwesenheit von NO ab, was eine begrenzte Wirksamkeit in Lungenregionen mit vermindertem NO-Spiegel bedeutet (
68
,
69
).Sildenafil wurde insbesondere auf seine Wirkung auf chronisch thromboembolische pulmonale Hypertonie (CTEPH) untersucht. In einer offenen, unkontrollierten Studie von Reichenberger et al. wurden 104 Patienten mit inoperabler CTEPH dreimal täglich mit Sildenafil in einer Dosierung von 50 mg behandelt. Die Ergebnisse zeigten eine signifikante Reduktion des pulmonalvaskulären Widerstandes (PVR) von einem Ausgangswert von 863 ± 38 dyn·s·cm −5 auf 759 ± 62 dyn·s·cm −5 nach drei Monaten ( p = 0,0002 vs. Ausgangswert), zusammen mit einer bemerkenswerten Erhöhung der 6-Minuten-Gehstrecke (6MWD) von 310 ± 11 m zu Beginn auf 361 ± 15 m nach drei Monaten ( p = 0,0001 vs. Ausgangswert) und 366 ± 18 m nach zwölf Monaten ( p = 0,0005 vs. Ausgangswert), was auf wesentliche hämodynamische und funktionelle Verbesserungen hindeutet (
70
).Während Sildenafil bei inoperabler CTEPH aufgrund des Fehlens randomisierter kontrollierter Studien (RCTs) oder Registerdaten, die seine Wirksamkeit belegen, außerhalb der Zulassung eingesetzt wird, hat sich die orale Kombinationstherapie mit PDE5-Hemmern und Endothelin-Rezeptorantagonisten als gängige Methode zur Behandlung von Patienten mit CTEPH mit schweren hämodynamischen Beeinträchtigungen etabliert (
71
). Diese Praxis unterstreicht die Integration verschiedener pharmakologischer Strategien zur Linderung der Symptome und Verbesserung der Lebensqualität der Patienten mit dieser herausfordernden Erkrankung.2.3.6 Löslicher Guanylatcyclase-StimulatorObwohl in zahlreichen randomisierten kontrollierten Studien die Wirksamkeit und Sicherheit verschiedener Behandlungen untersucht wurde, ist Riociguat das einzige Medikament, das von der FDA speziell sowohl für pulmonale Hypertonie (PH) als auch für chronisch thromboembolische pulmonale Hypertonie (CTEPH) zugelassen wurde. Riociguat, ein Stimulator der löslichen Guanylatcyclase (sGC), wird zur Behandlung von inoperabler CTEPH sowie persistierender/rezidivierender CTEPH nach pulmonaler Endarteriektomie (PEA) empfohlen und erhält IB-Empfehlungen (
2
,
72
). Dieses Medikament wirkt, indem es sGC direkt stimuliert, die cGMP-Werte unabhängig von Stickstoffmonoxid (NO) erhöht und die Empfindlichkeit der Gefäßwandzellen gegenüber endogenem NO steigert und so die Vasodilatation in schlecht belüfteten Lungenbereichen erleichtert (
73
).Erste Berichte von Ghofrani et al. aus dem Jahr 2010 zeigten, dass Riociguat die körperliche Leistungsfähigkeit bei Patienten mit CTEPH und PAH über einen Zeitraum von 12 Wochen verbessern und die Krankheitssymptome reduzieren konnte (
74
). Die PATENT-1-Studie bestätigte den Nutzen von Riociguat bei PAH-Patienten weiter und zeigte signifikante Verbesserungen der körperlichen Leistungsfähigkeit, der WHO-Funktionsklasse, des pulmonal-vaskulären Widerstands (PVR) und der NT-proBNP-Werte (
75
). Die langfristige Sicherheit und Wirksamkeit wurden in der PATENT-2-Studie bestätigt, in der es über ein Jahr hinweg zu anhaltenden Verbesserungen der funktionellen und körperlichen Leistungsfähigkeit kam (
76
).In der PATENT PLUS-Studie wurden die additiven Effekte von Riociguat und PDE5-Hemmern bei PAH-Patienten untersucht, die mit Sildenafil behandelt wurden. Zwischen den beiden Gruppen konnten keine signifikanten Unterschiede hinsichtlich der hämodynamischen Verbesserungen oder der körperlichen Leistungsfähigkeit festgestellt werden (
77
). In der CHEST-1-Studie an inoperablen CTEPH-Patienten wurden unter Riociguat-Behandlung ähnliche Verbesserungen der körperlichen Leistungsfähigkeit, der WHO-Funktionsklasse, des PVR und der NT-proBNP-Werte berichtet (
12
). Der langfristige Nutzen wurde in der CHEST-2-Studie bestätigt (
78
).Trotz dieser positiven Ergebnisse wurden in den Studien jedoch keine Parameter der Rechtsherzfunktionsstörung direkt ausgewertet, die für die Vorhersage des Überlebens entscheidend sind. Eine umfassende Analyse von Marra A. et al. mit Patienten aus den Studien PATENT-1, PATENT PLUS, EAS und CHEST zeigte im Verlauf einer 12-monatigen Riociguat-Therapie signifikante Reduktionen der rechtsventrikulären Dimensionen und Verbesserungen der TAPSE (
79
).Es ist wichtig zu beachten, dass es keine direkten Vergleichsstudien zwischen Riociguat und anderen medizinischen Therapien für CTEPH gibt, da Unterschiede im Studiendesign direkte Vergleiche erschweren. Die Studien CHEST-1, CTREPH und MERIT-1 hatten jeweils unterschiedliche Patientenpopulationen und Basistherapiezuteilungen, was zur Variabilität der Patientendemographie und der Schwere der Erkrankung beitrug (
6
,
12
,
13
,
54
).Riociguat ist die einzige von der FDA zugelassene Behandlung für inoperable CTEPH oder post-PEA-Rest-PH (
72
). Während eine orale Kombinationstherapie, einschließlich PDE5-Hemmern und Endothelin-Rezeptor-Antagonisten, bei CTEPH-Patienten mit schweren hämodynamischen Störungen weit verbreitet ist (
71
), bleibt die Rolle von Vasodilatatoren als präoperative Behandlung umstritten, da sie den chirurgischen Eingriff potenziell verzögern kann (
67
,
80
). Im Gegensatz dazu kann bei Patienten, bei denen eine BPA in Betracht gezogen wird, eine medikamentöse Vorbehandlung die Lungenhämodynamik und die Verfahrenssicherheit verbessern, obwohl diese Empfehlung auf Evidenz von sehr geringer Qualität beruht (
2
).
3 TherapiezieleDie Behandlungsziele bei CTEPH sind vielschichtig und konzentrieren sich auf die Verbesserung von Überleben, Symptomen und Lebensqualität sowie hämodynamischen Parametern. Das Erreichen eines Niedrigrisikostatus in der Risikobewertung der European Society of Cardiology (ESC) ist mit besseren Ergebnissen verbunden. Dabei werden Parameter wie Funktionsklasse, körperliche Belastbarkeit (6-Minuten-Gehstrecke oder kardiopulmonale Belastungstests), Biomarker (BNP- oder NT-proBNP-Werte) und rechtsventrikuläre Funktion im Echokardiogramm berücksichtigt. Eine Studie von Hoeper et al. ( 81 ) zeigte, dass Patienten, die ein Niedrigrisikoprofil erreichten oder aufrechterhielten, signifikant bessere Überlebensraten hatten als Patienten mit mittlerem oder hohem Risiko.Die Senkung des mittleren pulmonalarteriellen Drucks (mPAP) ist ein weiteres wichtiges Ziel bei der Behandlung von CTEPH. Erhöhter mPAP ist ein direkter hämodynamischer Marker für die Schwere der Erkrankung und wurde mit unerwünschten Ergebnissen in Zusammenhang gebracht. Eine chirurgische Behandlung, insbesondere eine pulmonale Endarteriektomie (PEA), kann den mPAP äußerst wirksam senken und die Hämodynamik verbessern. Studien haben gezeigt, dass eine erfolgreiche PEA zu einer Normalisierung oder nahezu Normalisierung des mPAP führen kann, was mit einer verbesserten Überlebensrate und einem besseren Funktionsstatus einhergeht ( 24 ). Darüber hinaus wurde der Einfluss des mPAP auf den Langzeitverlauf in mehreren Studien hervorgehoben. So fand eine Studie von Cannon et al. ( 82 ) heraus, dass der postoperative mPAP ein starker Prädiktor der Überlebensrate von CTEPH-Patienten ist.Da sowohl das Erreichen eines Niedrigrisikostatus als auch die Senkung des mPAP wichtig sind, sollte eine umfassende Behandlungsstrategie auf beides abzielen. Die ESC/ERS-Leitlinien für die Diagnose und Behandlung der pulmonalen Hypertonie ( 2 ) empfehlen, ein umfassendes Niedrigrisikoprofil anzustreben, das die Senkung des mPAP, eine funktionelle Verbesserung und Parameter zur Risikostratifizierung umfasst. Medizinische Therapien wie Riociguat bei inoperabler oder residualer CTEPH und interventionelle Behandlungen wie die Ballon-Pulmonalisangioplastie (BPA) spielen ebenfalls eine Rolle bei der Senkung des mPAP und der Verbesserung anderer Risikoparameter ( 68 ).Derzeit besteht kein Konsens über spezifische posttherapeutische Ziele für CTEPH-Patienten nach PEA/BPA oder medikamentöser Therapie. Das Erreichen einer besseren WHO-Funktionsklasse (I-II), die Normalisierung oder nahezu Normalisierung der hämodynamischen Parameter in Ruhe (Beurteilung durch RHC 3–6 Monate nach dem Eingriff) und die Verbesserung der Lebensqualität werden von Experten als primäre Ziele angesehen ( 78 ).Was das Absetzen von Medikamenten gegen pulmonale arterielle Hypertonie (PAH) nach Erreichen der Behandlungsziele mit BPA oder PEA betrifft, deuten Belege darauf hin, dass dies in bestimmten Fällen möglich sein könnte. Eine Studie von Sugimura et al. zeigte, dass einige Patienten nach erfolgreicher BPA ihre PAH-Medikamente reduzieren oder absetzen konnten, insbesondere wenn sie erhebliche Verbesserungen bei mPAP und anderen hämodynamischen Parametern erzielten ( 83 ). Die Entscheidung zum Absetzen von PAH-Medikamenten sollte jedoch individuell getroffen werden und die allgemeine Stabilität des Patienten und sein Ansprechen auf die Behandlung berücksichtigen.Zusammenfassend sollte das Ziel der CTEPH-Behandlung darin bestehen, einen Niedrigrisikostatus gemäß der ESC-Risikobewertung zu erreichen und aufrechtzuerhalten und gleichzeitig mPAP zu reduzieren. Dieser duale Ansatz wird durch Belege gestützt, die zeigen, dass beide Strategien eine verbesserte Überlebensrate und Lebensqualität aufweisen. Um die Ergebnisse bei CTEPH-Patienten zu optimieren, ist eine umfassende Behandlung mit chirurgischen, medizinischen und interventionellen Therapien unerlässlich.4 NachverfolgungUm die besten Ergebnisse für Patienten mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) zu erzielen, müssen sie fortlaufend in spezialisierten Zentren für die Behandlung von CTEPH betreut werden. Diese Zentren sollten über ein multidisziplinäres Team verfügen, das einen Chirurgen mit Erfahrung in der pulmonalen Endarteriektomie (PEA), einen Interventionisten für die Ballon-Pulmonalarterien-Angioplastie (BPA), einen Spezialisten für pulmonale Hypertonie (PH) und einen Thoraxradiologen umfasst, die alle in Zentren mit hohem PEA- und/oder BPA-Volumen ausgebildet wurden. Vor der Entlassung müssen die Patienten gründlich über die Wichtigkeit der Einhaltung der Medikamenteneinnahme und den Zeitplan für Nachuntersuchungen aufgeklärt werden. Dies ist besonders wichtig für Patienten mit mehreren Komorbiditäten, da eine sorgfältige Behandlung dieser zusätzlichen Gesundheitsprobleme ihre funktionelle Kapazität und allgemeine Lebensqualität deutlich verbessern kann.Obwohl es keine spezifische Empfehlung zur Risikobewertung bei Patienten mit inoperabler chronischer thromboembolischer pulmonaler Hypertonie (CTEPH) gibt, stellt d
Chronische thromboembolische pulmonale Hypertonie: die therapeutische Bewertung
Chronische thromboembolische pulmonale Hypertonie (CTEPH) ist eine schwere und komplexe Erkrankung, die sich aus einer nicht behandelten Lungenembolie entwickelt und zu einer fibrotischen Obstruktion der Lungenarterien, pulmonaler Hypertonie und potenzieller Rechtsherzinsuffizienz führt. Der Eckpfeiler der CTEPH-Behandlung ist ein vielschichtiger therapeutischer Ansatz, der auf das individuelle Patientenprofil zugeschnitten ist und die Heterogenität der Krankheit widerspiegelt. Dieser Bericht befasst sich mit den aktuellen Therapiestrategien für CTEPH, darunter chirurgische Lungenendarteriektomie (PEA), Ballon-Pulmonalangioplastie (BPA) und gezielte pharmakologische Behandlungen wie PDE5-Hemmer, Endothelin-Rezeptor-Antagonisten, sGC-Stimulatoren und Prostanoide. Eine lebenslange Antikoagulation wird ebenfalls als vorbeugende Strategie gegen wiederkehrende Thromboembolien hervorgehoben. Besonderer Wert wird auf die interdisziplinäre Natur der CTEPH-Behandlung gelegt, die eine Zusammenarbeit zwischen PEA-Chirurgen, BPA-Interventionisten, PH-Spezialisten und Thoraxradiologen erfordert, um eine umfassende Behandlungsplanung und -durchführung sicherzustellen. Die Überprüfung unterstreicht die Bedeutung der Auswahl einer geeigneten Behandlungsmethode auf der Grundlage der spezifischen Krankheitsmerkmale des Patienten und der sich entwickelnden Landschaft der CTEPH-Behandlung, mit dem Ziel, die Patientenergebnisse durch integrierte Behandlungsstrategien zu verbessern. 1 EinleitungPulmonale Hypertonie (PH) ist definiert als ein mittlerer pulmonalarterieller Druck (PAP) von größer oder gleich 20 mmHg, gemessen durch Rechtsherzkatheterisierung (RHC) ( 1 , 2 ).Die chronisch thromboembolische Lungenerkrankung (CTEPH), eine potenziell lebensbedrohliche Erkrankung, ist definiert als symptomatische pulmonale Hypertonie mit anhaltenden Lungendurchblutungsstörungen trotz adäquater Antikoagulation über 3–6 Monate ( 3 ) und stellt eine eigenständige Krankheitseinheit dar, die gemäß den ESC/ERS-Leitlinien 2022 ( 2 ) als pulmonale Hypertonie (PH) der Gruppe 4 klassifiziert wird .CTEPH ist eine seltene und unterdiagnostizierte Komplikation einer akuten Lungenembolie (APE) ( 4 ). Einige Merkmale der ursprünglichen Lungenembolie sind mit der Entwicklung von CTEPH assoziiert. Besonders wichtig ist, dass eine unprovozierte Lungenembolie, eine Diagnoseverzögerung von >2 Wochen und eine Funktionsstörung des rechten Ventrikels (RV) zum Zeitpunkt der Lungenembolie unabhängige Prädiktoren für CTEPH sind ( 5 ).Darüber hinaus ist die genaue Inzidenz von CTEPH nach einer dokumentierten und korrekt behandelten APE noch immer unbekannt.Die genaue Epidemiologie der CTEPH ist unbekannt; sie wird höchstwahrscheinlich weitgehend unterdiagnostiziert und daher unterbehandelt und zeigt, wie sehr Patienten von modernen multimodalen Behandlungskonzepten in Expertenzentren profitieren können.Chronische thromboembolische Lungenerkrankung (CTEPD) ist ein allgemeiner Begriff, der im jüngsten ERS-Statement zur chronischen thromboembolischen pulmonalen Hypertonie (CTEPH) ( 6 ) vorgeschlagen wurde, um symptomatische Patienten zu charakterisieren, die nach mindestens drei Monaten therapeutischer Antikoagulation in der Ventilations-/Perfusions-Lungenszintigraphie (V/Q) nicht übereinstimmende Perfusionsdefekte und in der Computertomographie-Pulmonalisangiographie (CTPA), konventionellen Pulmonalisangiographie (CPA) oder Magnetresonanztomographie (MRT) spezifische Anzeichen chronischer organisierter Gerinnsel aufweisen. Einige dieser Patienten weisen im Ruhezustand keine pulmonale Hypertonie (PH) auf ( 2 ), während bei der Mehrheit eine PH im Ruhezustand vorliegt, entsprechend der Definition der CTEPH (Gruppe 4 der aktualisierten klinischen Klassifikation der PH) ( 1 ).Der natürliche Verlauf von CTEPH ist komplex. Das aktuelle Verständnis der Pathophysiologie von CTEPH hat sich in letzter Zeit über eine einfache pulmonalvaskuläre Erkrankung hinaus entwickelt, die durch eine intravaskuläre mechanische Obstruktion aufgrund von organisiertem chronischem fibrotischem Material aus nicht aufgelösten Blutgerinnseln verursacht wird. Tatsächlich könnte es sich um eine viel komplexere Erkrankung handeln, die eine proximale und distalere Obstruktion der Lungenarterien umfasst, die mit einer Umgestaltung der muskulären Lungenarterien, Kapillaren und Venolen einhergeht ( 7 ).Heute ist es allgemein anerkannt, dass pulmonale Gefäßumbauprozesse zu einer signifikanten pulmonalen Mikrovaskulopathie führen können, die wiederum bei der Entwicklung eines progressiven Anstiegs des pulmonalen Gefäßwiderstands und in der Folge bei der Entstehung von Funktionsstörungen der rechten Herzhälfte und symptomatischer CTEPH eine Rolle spielt ( 8 , 9 ). Die Prozesse, die zur Persistenz organisierter Gerinnsel nach einer APE führen, sind noch nicht vollständig verstanden, auch wenn mehrere Risikofaktoren identifiziert wurden.Die Risikofaktoren für CTEPH scheinen sich von denen für APE zu unterscheiden, wie z. B. Immobilisierung oder eine kürzlich erfolgte Operation. Zu den potenziellen Risikofaktoren für CTEPH gehören bestimmte chronische Erkrankungen (z. B. permanente intravaskuläre Geräte, entzündliche Darmerkrankungen, Autoimmunerkrankungen, Hypothyreose, Splenektomie und Malignität), Thrombophilie und genetische Veranlagung.Zur Diagnostik können Echokardiographie, CT und Ventilations-Perfusions-Scans (V/Q-Scans) eingesetzt werden, um den Verdacht auf PH zu bestätigen. Das Vorliegen einer V/Q-Inkongruenz im Rahmen einer PH sollte eine weitere Untersuchung mittels RHC und Pulmonalisangiographie nach sich ziehen. Jedes Bildgebungsverfahren hat seine Funktion; daher ist eine umfassende Untersuchung mittels multimodaler Bildgebung für die richtige Diagnose und Behandlung von Patienten mit CTEPH entscheidend. Die Seltenheit der Erkrankung, unspezifische Symptome und Anzeichen sowie die mangelnde Kenntnis der Ärzte über CTEPH (einschließlich des Zeitpunkts des Verdachts und der Art und Weise, wie sie zu beurteilen ist) erschweren eine rechtzeitige Diagnose ( 10 ).Daher werden viele Patienten trotz wirksamer Therapie [pulmonale Endarteriektomie (PEA)] erst im Spätstadium der Erkrankung diagnostiziert, wenn die distale PA-Obstruktion und die Mikrovaskulopathie bereits fortgeschritten sind. Patienten mit fortgeschrittener Erkrankung kommen für eine PEA nicht in Frage, da mit diesem Verfahren nur proximale Läsionen behandelt werden können.Glücklicherweise wurden parallel zu den Fortschritten bei den Diagnose- und Operationstechniken für Patienten mit operablem CTEPH auch erhebliche therapeutische Fortschritte eingeführt. Dazu gehören die gezielte medizinische Therapie und die Ballon-Pulmonalisangioplastie (BPA) für Patienten, bei denen aufgrund der distalen Lage der chronischen thromboembolischen Obstruktion, der Schwere der hämodynamischen Beeinträchtigung, des Vorhandenseins schwerer Komorbiditäten oder persönlicher Präferenz eine inoperable CTEPH angenommen wird ( 11 – 14 ).Daher sind für optimale Patientenergebnisse ein hohes Verdachtspotenzial und eine rechtzeitige Diagnose mithilfe multimodaler Bildgebungsverfahren zwingend erforderlich.Ziel der vorliegenden Übersicht ist es, das Bewusstsein für diese seltene Erkrankung zu schärfen und neuere Bemühungen zur Entwicklung von Behandlungslösungen für verschiedene Phänotypen der CTEPH darzulegen.
2 TherapieDie moderne Therapiestrategie für chronisch thromboembolische pulmonale Hypertonie (CTEPH) verfolgt einen umfassenden, multimodalen Ansatz, der pulmonale Endarteriektomie (PEA), pulmonale Ballonangioplastie (BPA) und pharmakologische Behandlungen integriert, um die vielfältigen anatomischen Herausforderungen durch proximale, distale und mikrovaskuläre Läsionen zu bewältigen ( Abbildung 1 ). PEA stellt eine potenziell kurative Option dar, ihre Anwendbarkeit ist jedoch durch das Operationsrisiko oder die Unzugänglichkeit der Krankheit bei bestimmten Patienten beschränkt, sodass alternative Eingriffe wie BPA und medikamentöse Therapie erforderlich sind. In Ausnahmefällen, etwa bei Patienten mit gemischten anatomischen Läsionen (operativ erreichbare Läsionen in einer Lunge und inoperable Läsionen in der anderen Lunge), kann ein kombinierter Ansatz mit BPA (vor oder gleichzeitig mit der Operation) und PEA von Nutzen sein, um das Operationsrisiko zu senken und das Endergebnis zu verbessern ( 2 ). Abildung 1
2.3 Medikamentöse TherapieDie medikamentöse Therapie spielt bei der Behandlung von chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) eine entscheidende Rolle, insbesondere bei der Behandlung der mikrovaskulären Erkrankung sowie bei Patienten, die als inoperabel gelten oder an anhaltender oder wiederkehrender PH nach pulmonaler Endarteriektomie (PEA) leiden. Die Grundlage der medikamentösen Therapie bei CTEPH umfasst eine lebenslange Antikoagulation zur Vorbeugung weiterer thromboembolischer Ereignisse sowie pulmonale Vasodilatatoren zur Behandlung der PH. Zur Behandlung einer Volumenüberlastung können Diuretika verschrieben werden, und bei Bedarf wird zusätzlicher Sauerstoff zur Korrektur der Hypoxämie eingesetzt.Drei bedeutende klinische Studien haben wesentlich zur Evidenzbasis für die pharmakologische Behandlung von CTEPH beigetragen: CHEST-1-Studie (Riociguat), CTREPH-Studie (Treprostinil) und MERIT-1-Studie (Macitentan), die später ausführlicher besprochen werden ( Tabelle 5 ). Tbelle 5
3 TherapiezieleDie Behandlungsziele bei CTEPH sind vielschichtig und konzentrieren sich auf die Verbesserung von Überleben, Symptomen und Lebensqualität sowie hämodynamischen Parametern. Das Erreichen eines Niedrigrisikostatus in der Risikobewertung der European Society of Cardiology (ESC) ist mit besseren Ergebnissen verbunden. Dabei werden Parameter wie Funktionsklasse, körperliche Belastbarkeit (6-Minuten-Gehstrecke oder kardiopulmonale Belastungstests), Biomarker (BNP- oder NT-proBNP-Werte) und rechtsventrikuläre Funktion im Echokardiogramm berücksichtigt. Eine Studie von Hoeper et al. ( 81 ) zeigte, dass Patienten, die ein Niedrigrisikoprofil erreichten oder aufrechterhielten, signifikant bessere Überlebensraten hatten als Patienten mit mittlerem oder hohem Risiko.Die Senkung des mittleren pulmonalarteriellen Drucks (mPAP) ist ein weiteres wichtiges Ziel bei der Behandlung von CTEPH. Erhöhter mPAP ist ein direkter hämodynamischer Marker für die Schwere der Erkrankung und wurde mit unerwünschten Ergebnissen in Zusammenhang gebracht. Eine chirurgische Behandlung, insbesondere eine pulmonale Endarteriektomie (PEA), kann den mPAP äußerst wirksam senken und die Hämodynamik verbessern. Studien haben gezeigt, dass eine erfolgreiche PEA zu einer Normalisierung oder nahezu Normalisierung des mPAP führen kann, was mit einer verbesserten Überlebensrate und einem besseren Funktionsstatus einhergeht ( 24 ). Darüber hinaus wurde der Einfluss des mPAP auf den Langzeitverlauf in mehreren Studien hervorgehoben. So fand eine Studie von Cannon et al. ( 82 ) heraus, dass der postoperative mPAP ein starker Prädiktor der Überlebensrate von CTEPH-Patienten ist.Da sowohl das Erreichen eines Niedrigrisikostatus als auch die Senkung des mPAP wichtig sind, sollte eine umfassende Behandlungsstrategie auf beides abzielen. Die ESC/ERS-Leitlinien für die Diagnose und Behandlung der pulmonalen Hypertonie ( 2 ) empfehlen, ein umfassendes Niedrigrisikoprofil anzustreben, das die Senkung des mPAP, eine funktionelle Verbesserung und Parameter zur Risikostratifizierung umfasst. Medizinische Therapien wie Riociguat bei inoperabler oder residualer CTEPH und interventionelle Behandlungen wie die Ballon-Pulmonalisangioplastie (BPA) spielen ebenfalls eine Rolle bei der Senkung des mPAP und der Verbesserung anderer Risikoparameter ( 68 ).Derzeit besteht kein Konsens über spezifische posttherapeutische Ziele für CTEPH-Patienten nach PEA/BPA oder medikamentöser Therapie. Das Erreichen einer besseren WHO-Funktionsklasse (I-II), die Normalisierung oder nahezu Normalisierung der hämodynamischen Parameter in Ruhe (Beurteilung durch RHC 3–6 Monate nach dem Eingriff) und die Verbesserung der Lebensqualität werden von Experten als primäre Ziele angesehen ( 78 ).Was das Absetzen von Medikamenten gegen pulmonale arterielle Hypertonie (PAH) nach Erreichen der Behandlungsziele mit BPA oder PEA betrifft, deuten Belege darauf hin, dass dies in bestimmten Fällen möglich sein könnte. Eine Studie von Sugimura et al. zeigte, dass einige Patienten nach erfolgreicher BPA ihre PAH-Medikamente reduzieren oder absetzen konnten, insbesondere wenn sie erhebliche Verbesserungen bei mPAP und anderen hämodynamischen Parametern erzielten ( 83 ). Die Entscheidung zum Absetzen von PAH-Medikamenten sollte jedoch individuell getroffen werden und die allgemeine Stabilität des Patienten und sein Ansprechen auf die Behandlung berücksichtigen.Zusammenfassend sollte das Ziel der CTEPH-Behandlung darin bestehen, einen Niedrigrisikostatus gemäß der ESC-Risikobewertung zu erreichen und aufrechtzuerhalten und gleichzeitig mPAP zu reduzieren. Dieser duale Ansatz wird durch Belege gestützt, die zeigen, dass beide Strategien eine verbesserte Überlebensrate und Lebensqualität aufweisen. Um die Ergebnisse bei CTEPH-Patienten zu optimieren, ist eine umfassende Behandlung mit chirurgischen, medizinischen und interventionellen Therapien unerlässlich.4 NachverfolgungUm die besten Ergebnisse für Patienten mit chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) zu erzielen, müssen sie fortlaufend in spezialisierten Zentren für die Behandlung von CTEPH betreut werden. Diese Zentren sollten über ein multidisziplinäres Team verfügen, das einen Chirurgen mit Erfahrung in der pulmonalen Endarteriektomie (PEA), einen Interventionisten für die Ballon-Pulmonalarterien-Angioplastie (BPA), einen Spezialisten für pulmonale Hypertonie (PH) und einen Thoraxradiologen umfasst, die alle in Zentren mit hohem PEA- und/oder BPA-Volumen ausgebildet wurden. Vor der Entlassung müssen die Patienten gründlich über die Wichtigkeit der Einhaltung der Medikamenteneinnahme und den Zeitplan für Nachuntersuchungen aufgeklärt werden. Dies ist besonders wichtig für Patienten mit mehreren Komorbiditäten, da eine sorgfältige Behandlung dieser zusätzlichen Gesundheitsprobleme ihre funktionelle Kapazität und allgemeine Lebensqualität deutlich verbessern kann.Obwohl es keine spezifische Empfehlung zur Risikobewertung bei Patienten mit inoperabler chronischer thromboembolischer pulmonaler Hypertonie (CTEPH) gibt, stellt d
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.