
- Beiträge: 1757
Sidebar
CTEPH, Hintergründe
24 Nov 2024 21:30 #2282
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
CTEPH, Hintergründe wurde erstellt von danny
www.frontiersin.org/journals/cardiovascu...vm.2024.1439402/full
Chronische thromboembolische pulmonale Hypertonie (CTEPH) stellt aufgrund ihrer komplexen und häufig unspezifischen klinischen Erscheinungsformen eine erhebliche diagnostische Herausforderung dar. Dieser Bericht skizziert einen umfassenden Ansatz zur diagnostischen Beurteilung von CTEPH und betont, wie wichtig ein hoher Verdachtsindex bei Patienten mit unerklärlicher Dyspnoe oder anhaltenden Symptomen nach akuter Lungenembolie ist. Wir erörtern die zentrale Rolle multimodaler Bildgebung, darunter Echokardiografie, Ventilations-/Perfusionsscans, CT-Pulmonalisangiografie und Magnetresonanztomographie bei der Identifizierung und Bestätigung von CTEPH. Außerdem unterstreicht der Bericht die wesentliche Funktion der Rechtsherzkatheterisierung bei der Validierung der hämodynamischen Parameter, die auf CTEPH hinweisen, und der Sicherstellung der definitiven Diagnose. Fortschritte bei Diagnosetechnologien und die Integration eines multidisziplinären Ansatzes sind für eine rechtzeitige und genaue Diagnose von CTEPH von entscheidender Bedeutung, da sie frühe therapeutische Eingriffe ermöglichen und die Patientenergebnisse verbessern. Ziel dieses Manuskripts ist es, Klinikärzte mit dem Wissen und den Werkzeugen auszustatten, die für einen effizienten Diagnoseablauf bei CTEPH erforderlich sind, und das Bewusstsein und Verständnis für diese potenziell behandelbare Ursache der pulmonalen Hypertonie zu fördern.1 EinleitungPulmonale Hypertonie (PH) ist definiert durch einen erhöhten mittleren pulmonalarteriellen Druck (mPAP), gemessen durch eine Rechtsherzkatheterisierung (RHC) und kann durch verschiedene Erkrankungen des Lungenkreislaufs verursacht werden ( 1 ). Sie ist oft mit Herz-Kreislauf- und Atemwegserkrankungen verbunden und wird durch Zellproliferation und Fibrose in den kleinen Lungenarterien verursacht, was zu einem erhöhten pulmonalvaskulären Widerstand (PVR) und schließlich zu einer Rechtsherzinsuffizienz führt, die die Hauptursache für Morbidität und Mortalität bei PAH ist ( 2 ). Die Behandlung von PH erfordert einen multidisziplinären Ansatz, bei dem die Zusammenarbeit zwischen Patienten und medizinischem Personal im Vordergrund steht ( 3 ).Leichte Erhöhungen des pulmonalarteriellen Drucks sind signifikante Prädiktoren der Mortalität und unterstreichen die Notwendigkeit einer frühzeitigen Erkennung und Behandlung der PH ( 4 ).Chronische thromboembolische pulmonale Hypertonie (CTEPH) ist eine schwere Form der PH, die durch intraluminale Thrombosen, fibröse Gewebe und Obliteration der Pulmonalarterien gekennzeichnet ist ( 5 ). Die genauen molekularen Auslöser für die umfassenden Umbauvorgänge und die Fibrose bei CTEPH sind unbekannt. Man geht davon aus, dass die Erkrankung durch nicht aufgelöstes thromboembolisches Material in den Pulmonalarterien verursacht wird, wo ein niedriger Sauerstoffpartialdruck die Transkription von Genen fördert, die an Gerinnung, Angiogenese, Entzündung und Fibrose beteiligt sind. Diese Erkrankung ist durch ein proliferatives und entzündliches Zellumfeld gekennzeichnet, das zu Fibrose, Narbenbildung und okklusiven Läsionen führt ( 6 ). Bei der Behandlung dieser Patienten ist eine Risikostratifizierung von entscheidender Bedeutung ( 7 ).In diesem Bericht werden die grundlegenden Mechanismen von pulmonal-hypertensiven Erkrankungen erörtert, auf den aktuellen Kenntnisstand zur Pathobiologie von CTEPH eingegangen und Diagnoseinstrumente sowie Behandlungsstrategien zur Erzielung optimaler Behandlungsergebnisse für die Patienten dargelegt. 2 Definitionen und KlassifikationenIm Jahr 2009 analysierten Kovacs et al. RHC-Daten von gesunden Personen, um Normalwerte für den mittleren pulmonalarteriellen Druck (mPAP) in Ruhe und unter Belastung festzulegen. Sie überprüften Daten von 1.187 gesunden Personen aus 47 Studien und fanden einen Ruhe-mPAP von 14,0 ± 3,3 mmHg, ein Wert, der für alle Geschlechter und Ethnien gleich ist und nur minimalen Einfluss von Alter und Körperhaltung hat. Ein Ruhe-mPAP von über 20 mmHg – abgeleitet durch die Addition zweier Standardabweichungen – wurde als Überschreitung der normalen Obergrenze (über dem 97,5. Perzentil) identifiziert und korrelierte mit einem erhöhten Mortalitätsrisiko aller Ursachen. Der durch Belastung bedingte mPAP liegt oft über 30 mmHg, insbesondere bei älteren Erwachsenen, was die Festlegung von Standardwerten für den mPAP unter Belastung erschwert ( 3 , 4 , 8 , 9 ). Die ERS Task Force definiert PH unter Belastung vorläufig als einen mPAP von über 30 mmHg bei einem totalen Lungenwiderstand (TPR) von über 3 Wood-Einheiten (WU) bei maximaler Belastung, wobei alternativ eine mPAP/Herzleistung (CO)-Steigung von über 3 WU als Grenzwert vorgeschlagen wird. Die Steigungen von mPAP/CO und pulmonalarteriellem Verschlussdruck (PAWP)/CO sind entscheidend für die Beurteilung der Lungenzirkulation unter Belastung, da sie stark altersabhängig sind und bei unterschiedlichen Belastungen gleichbleibend sind. Eine PAWP/CO-Steigung von über 2 WU wird mit negativen kardiovaskulären Folgen in Verbindung gebracht und hilft bei der Unterscheidung zwischen prä- und postkapillären Ursachen von PH unter Belastung ( 10 ). Erhöhte mPAP-Werte unter bestimmten physiologischen Zuständen sind allein kein Hinweis auf eine pulmonalarterielle Gefäßerkrankung. Der PVR ist für die Prognose und die klinische Entscheidungsfindung bei pulmonaler arterieller Hypertonie (PAH) von entscheidender Bedeutung. Die vorliegenden Daten deuten darauf hin, dass die obere Normgrenze und der niedrigste prognostisch relevante PVR-Schwellenwert bei etwa 2 WU liegen ( 11 – 13 ).Die ESC/ERS-Leitlinien 2022 zur Diagnose und Behandlung von pulmonaler Hypertonie klassifizieren PH basierend auf den mPAP-, PAWP- und PVR-Werten in drei Haupttypen:• Präkapilläre pulmonale Hypertonie: mPAP >20 mmHg, PAWP <15 mmHg, PVR >2 WU.• Postkapilläre pulmonale Hypertonie (IpcPH): mPAP >20 mmHg, PAWP >15 mmHg, PVR <2 WU.• Kombinierte pulmonale Hypertonie (CpcPH): mPAP >20 mmHg, PAWP >15 mmHg, PVR >2 WU. Darüber hinaus sind die Belastungs-PH und die nicht klassifizierte PH anerkannt. Zu letzterer zählen Patienten mit mPAP >20 mmHg, PVR <2 WU und PAWP <15 mmHg, oft in Verbindung mit einer erhöhten Lungendurchblutung und Erkrankungen wie angeborenen Herzfehlern, Lebererkrankungen oder Lungenerkrankungen, was weitere ätiologische Studien und klinische Nachuntersuchungen erforderlich macht ( 3 ).PH wird anhand pathophysiologischer, klinischer und therapeutischer Aspekte in fünf Gruppen eingeteilt: • Gruppe 1: Pulmonale arterielle Hypertonie (PAH) • Gruppe 2: PH aufgrund einer linksseitigen Herzerkrankung • Gruppe 3: PH aufgrund von Lungenerkrankungen oder Hypoxie • Gruppe 4: PH in Verbindung mit einer Obstruktion der Lungenarterien (kann auf CTEPH, thrombotische Mikroangiopathie durch Lungentumoren oder andere Ursachen für eine Obstruktion der Lungenarterien zurückzuführen sein, beispielsweise Sarkome (hoch- oder mittelgradig oder Angiosarkom), andere bösartige Tumoren (z. B. Nierenkarzinom, Uteruskarzinom, Keimzelltumoren der Hoden) oder nicht bösartige Tumoren (z. B. Uterusleiomyom), Arteriitis ohne Bindegewebserkrankung, angeborene Lungenarterienstenosen und Echinokokkose) ( 14 ) • Gruppe 5: PH mit unklaren oder multifaktoriellen Mechanismen Unabhängig von der zugrundeliegenden Ursache ist die Entwicklung einer PH mit einer Verschlechterung der Symptome und einem erhöhten Sterberisiko verbunden ( 14 ).Zur pulmonal-arteriellen Hypertonie (PAH) der Gruppe 1 gehören: • Idiopathische PAH • Erbliche PAH • Arzneimittel- und toxinbedingte PAH • PAH verbunden mit:○ Bindegewebserkrankungen○ HIV-Infektion○ Portale Hypertonie○ Angeborene Herzerkrankungen○ Bilharziose○ Chronische hämolytische Anämie Diese Klassifikation orientiert sich an den Leitlinien der ESC/ERS und gewährleistet eine umfassende Abdeckung der verschiedenen Ursachen der PAH ( Ergänzende Tabelle 1 ).Gruppe 2 umfasst die postkapilläre PH, entweder isoliert oder kombiniert mit einer präkapillären Komponente. Sie tritt häufig bei Herzinsuffizienz mit erhaltener oder reduzierter Ejektionsfraktion auf und betrifft über 50 % der entsprechenden Patienten ( 15 , 16 ). In beiden Patientengruppen ist das Mortalitätsrisiko bei Funktionsstörungen des rechten Ventrikels doppelt so hoch. Ebenso entwickeln 50–70 % der Patienten mit schwerer Aortenstenose eine PH, wodurch sich ihr Mortalitätsrisiko verdoppelt ( 17 , 18 ). Ebenso wird eine PH bei 60–70 % der Patienten mit schwerer, symptomatischer Mitralklappenerkrankung beobachtet, wobei die Prävalenz mit zunehmender Schwere der linksseitigen Klappenerkrankungen zunimmt ( 19 ).Patienten mit Herzinsuffizienz und erhaltener Ejektionsfraktion (HFpEF), die in erster Linie an Belastungsdyspnoe leiden, können in Ruhe normale Füllungsdrücke, aber bei leichter Belastung deutlich erhöhte Drücke aufweisen ( 20 ). Diese Patientengruppe, bei der es sich oft um ältere Menschen handelt, zeigt während der Belastung eine Abnahme des Herzzeitvolumens aufgrund einer verringerten ventrikulären Compliance und Relaxation mit dem Alter. Belastungstests können frühe Anzeichen einer HFpEF aufdecken, selbst bei normalen Ruhewerten des pulmonalkapillären Verschlussdrucks (PCWP) (<15 mmHg). Bemerkenswerterweise weisen 30 % der gesunden Personen über 60 Jahre bei Belastungstests PCWP-Werte über 25 mmHg auf, was auf den potenziellen Nutzen altersangepasster PCWP-Diagnoseschwellenwerte für eine verbesserte Diagnosegenauigkeit hindeutet ( 21 ).Bei Patienten mit isolierter postkapillärer PH aufgrund einer Gefäßstauung infolge einer Linksherzerkrankung können frühe Interventionen wie die Stabilisierung des intravaskulären Volumens oder die Behebung des primären Defekts die PH oft rückgängig machen, bevor es zu signifikanten Gefäßumbauvorgängen und einem erhöhten PVR kommt ( 22 ). Dies unterstreicht die dringende Notwendigkeit von Strategien, die auf eine frühe Erkennung und Diagnose der PH abzielen, um irreversible Gefäßveränderungen, die zu erhöhtem mPAP führen, zu verhindern.In Gruppe 3 ist die PH typischerweise leicht ausgeprägt, beeinträchtigt die Patienten jedoch erheblich, indem sie die Symptome verschlimmern, die körperliche Belastbarkeit verringern, die Zahl der Krankenhauseinweisungen erhöhen und die Sterblichkeit im Vergleich zu Patienten ohne PH erhöhen ( 14 ).Gruppe 4 konzentriert sich auf CTEPH, eine Erkrankung, die durch Symptome gekennzeichnet ist, die durch die Verstopfung der Lungenarterien durch chronische fibrotische Gerinnsel entstehen. Das Fortbestehen dieser Thromben und die daraus resultierende mikrovaskuläre Arteriopathie erhöhen den pulmonalen Gefäßwiderstand, was zu PH und Rechtsherzversagen führt. Einzigartig ist, dass CTEPH potenziell durch eine Lungenendarteriektomie (PEA) heilbar ist. Für die Diagnose ist eine Rechtsherzkatheteruntersuchung erforderlich, um Folgendes zu bestätigen:1. mPAP > 20 mmHg, PAWP ≤ 15 mmHg in Ruhe und PVR > 2 WU;2. Positive Befunde in der Lungenbildgebung, wie etwa Nichtübereinstimmungen bei der Ventilations-/Perfusionsszintigraphie (V/Q), erhebliche Perfusionsdefekte bei der Einzelphotonen-Emissionscomputertomographie (SPECT), Lungengefäßanomalien bei der CT/MRT oder Lungenangiographie, die auf chronische Verschlüsse hinweisen;3. Bestätigung dieser Zustände nach mindestens drei Monaten unter effektiver Antikoagulation, um eine Unterscheidung von einer subakuten Lungenembolie zu ermöglichen ( 3 , 23 – 26 ).
CTEPH und Chronische Thromboembolische Erkrankung (CTED) sind Begriffe, die symptomatische Personen mit chronischen Pulmonalarterienverschlüssen anhand des Vorhandenseins oder Fehlens von PH in Ruhe unterscheiden ( 24 ). CTED beschreibt Patienten mit mPAP <25 mmHg, anhaltenden Gefäßverschlüssen, Belastungsintoleranz und erheblichen Auswirkungen auf Symptom und Lebensqualität, denen eine PH in Ruhe fehlt. Trotz ähnlicher Symptome wie bei CTEPH können CTED-Patienten ähnliche chirurgische oder interventionelle Behandlungen erhalten. Ihre Belastungseinschränkung rührt von der belastungsbedingten PH und der Totraumventilation her ( 27 ). Die neuen mPAP- und PVR-Kriterien der Leitlinien von 2022 haben zu einer Neuklassifizierung einiger CTED-Patienten als CTEPH geführt und den Begriff „chronische thromboembolische Lungenerkrankung“ (CTEPD) eingeführt, um symptomatische Patienten mit Durchblutungsstörungen und chronischen Gerinnselzeichen unabhängig von der Ruhe-PH zu umfassen ( 24 ). Der Übergang von CTEPD-Patienten zu CTEPH und die Faktoren, die ihre Prognose beeinflussen, bleiben weiterhin Gegenstand eingehender Untersuchungen ( 25 , 28 ).Zur Gruppe 5 zählen Erkrankungen, die aufgrund vielfältiger und komplexer Mechanismen häufig mit PH in Zusammenhang stehen; hierzu zählen etwa chronisches Nierenversagen und Sarkoidose, die durch unterschiedliche pathophysiologische Ursachen charakterisiert sind ( 14 ).Bei belastungsbedingter PAH sind die mPAP/CO- und PAWP/CO-Steigungen entscheidend, da sie unabhängig von der Belastung Erkenntnisse liefern, aber stark altersabhängig sind. Eine mPAP/CO-Steigung von >3 mmHg/l/min zwischen Ruhe und Belastung ist für die Diagnose unverzichtbar, da höhere Steigungen auf eine schlechtere Überlebenschance bei allen kardiopulmonalen Erkrankungen hinweisen. Eine PAWP/CO-Steigung von >2 WU ist mit kardiovaskulären Risiken verbunden und hilft bei der Unterscheidung zwischen prä- und postkapillären PH-Ursachen bei körperlicher Belastung ( 10 ) ( Abbildung 1 ).ng 13 Epidemiologie und RisikofaktorenDie Prävalenz der PAH, kategorisiert als PH der Gruppe 1, beträgt etwa 25 Fälle pro 1 Million Einwohner, mit einer jährlichen Inzidenz von etwa fünf Fällen pro 1 Million Einwohner ( 29 , 30 ). Das mediane Alter bei Diagnose in klinischen Studien und Registern beträgt etwa 53 Jahre ( 31 ).Studien für Gruppe 4 zeigen, dass 1,0–8,8 % der Patienten, die eine akute Lungenembolie (PE) überleben, an CTEPH erkranken können, wobei die Inzidenz laut Chausheva et al. ( 5 ) bei etwa 3 % liegt. Die von Registern in Spanien und Großbritannien gemeldeten jährlichen Inzidenzraten betragen 0,9 bzw. 1,75 pro Million ( 32 , 33 ). Deutsche Daten deuten auf eine Inzidenz von 4,0 pro Million Erwachsene pro Jahr hin ( 14 ), die im Jahr 2016 auf 5,7 anstieg, während Frankreich 5–6 Fälle pro Million Erwachsene meldet ( 34 ). Die Registerdaten variieren und deuten auf eine CTEPH-Inzidenz und -Prävalenz von 2–6 bzw. 26–38 Fällen pro Million Erwachsene hin ( 35 – 37 ). Die steigende Zahl diagnostizierter CTEPH-Fälle ist auf ein besseres Verständnis der Krankheit und aktives Screening zurückzuführen, insbesondere bei Patienten, die nach einer PE symptomatisch bleiben oder ein Risiko für CTEPH haben ( 24 , 34 , 38 ). Die weltweite Prävalenz und Inzidenz von CTEPH sind weiterhin unklar, so dass für genaue Definitionen weitere Forschung erforderlich ist ( Abbildung 1 ) ( 28 ).In Europa und den USA beträgt die durchschnittliche Inzidenz von CTEPH etwa 4 %, im Gegensatz zu bis zu 14 % in Japan ( 15 , 39 ), wo die Lungenembolierate wesentlich niedriger ist. In Europa sind beide Geschlechter gleichermaßen von der Krankheit betroffen, während in Japan Frauen überwiegen ( 5 ). Das europäische CTEPH-Register zeigt, dass bei Frauen bestimmte kardiovaskuläre Risikofaktoren weniger häufig auftreten, jedoch Übergewicht, Krebs und Schilddrüsenerkrankungen häufiger auftreten ( 26 ). Bemerkenswerterweise haben 50–75 % der CTEPH-Patienten eine dokumentierte Vorgeschichte einer akuten Lungenembolie, während dieser Wert bei japanischen Patienten auf 15 % sinkt, was auf einen bestimmten CTEPH-Phänotyp hindeutet ( 24 ).Alle Metaanalysen berichten von einer CTEPH-Inzidenz von 2,7–3 %, wobei etwa 30 % der Patienten eine CTED entwickeln, eine symptomatische Restvaskulopathie ohne PH ( 23 , 34 , 35 ). In der FOCUS-Studie wurde insbesondere eine Inzidenz von Beeinträchtigungen nach einer Lungenembolie von 16 % berichtet, wobei CTEPH bei 2,3 % der Patienten auftrat ( 40 ). Es wurde auch berichtet, dass bis zu 20 % der Patienten nach einer Lungenembolie irgendeine Form von Beeinträchtigung erfahren können, was die Notwendigkeit einer aufmerksamen Überwachung und eines aktiven Screenings bei diesen Patienten unterstreicht, um langfristige Komplikationen wirksam behandeln und eindämmen zu können ( 40 ).Zu den Risikofaktoren für CTEPH zählen venöse Thromboembolien (VTE) in der Vorgeschichte, insbesondere wiederkehrende VTE, erhöhter pulmonalarterieller Druck nach Lungenembolie, maligne Erkrankungen in der Vorgeschichte, Splenektomie, Staphylokokkeninfektion, Blutgruppen außer O, Antiphospholipid-Antikörper, Lupusantikoagulans und permanente intravaskuläre Geräte. Weitere Risikofaktoren sind entzündliche Darmerkrankungen, Polycythaemia vera, essentielle Thrombozythämie, Hypothyreose, erhöhte Werte von Faktor VIII und von-Willebrand-Faktor, Fibrinogen-Polymorphismus und möglicherweise Faktor V Leiden, obwohl die Rolle des Letzteren noch umstritten ist ( 3 , 24 , 40 – 42 ). Demografische Merkmale und das Vorhandensein von chronischen entzündlichen/Autoimmunerkrankungen oder hämatologischen Erkrankungen spielen ebenfalls eine wichtige Rolle bei der Entstehung der Erkrankung ( 43 , 44 ) ( Tabelle 1 ).
CTEPH ist gekennzeichnet durch einen prothrombotischen Zustand und abnorme Fibrinogenmoleküle, die einer physiologischen Thrombolyse widerstehen, was auf eine beeinträchtigte Auflösung des anfänglichen embolischen Ereignisses hinweist. Eine Splenektomie ist insbesondere mit distalen CTEPH-Typen assoziiert und erschwert PEA-Verfahren ( 44 ). Eine Vorgeschichte infizierter ventrikuloatrialer Shunts und Splenektomien ist nicht nur mit CTEPH, sondern auch mit erheblichen Entzündungen und einem erhöhten VTE-Risiko verbunden ( 41 ). Darüber hinaus kann CTEPH durch eine postkapilläre Krankheitskomponente kompliziert werden, was die Bedeutung der Erkennung klinischer Risikofaktoren für PH im Zusammenhang mit Linksherzerkrankungen unterstreicht ( 47 ).4 PathophysiologieCTEPH ist durch vaskuläre Umbauprozesse in der Lunge gekennzeichnet, die sowohl verschlossene als auch nicht verschlossene kleine Gefäße betreffen, was zu hohen Morbiditäts- und Mortalitätsraten führt, insbesondere aufgrund der Progression zu einer Rechtsherzinsuffizienz, wenn die Erkrankung unbehandelt bleibt ( 24 , 48 , 49 ). CTEPH ist eine seltene Spätkomplikation einer APE, die auch nach mehr als drei Monaten Antikoagulationstherapie nicht abklingt und typischerweise auf eine venöse Thromboembolie zurückzuführen ist ( 50 ). Etwa 4 % der APE-Fälle entwickeln sich innerhalb von 2 Jahren zu CTEPH ( 51 ), wobei 75 % der CTEPH-Patienten eine APE-Anamnese haben ( 52 ).Der Grund, warum manche Menschen nach einer APE eine CTEPH entwickeln, ist weiterhin unklar, insbesondere angesichts der Tatsache, dass sich die meisten Gerinnsel unter Antikoagulation auflösen. CTEPH-Thromben sind faserig und weisen kollagene und entzündliche Komponenten auf, was manchmal zur Verkalkung führt ( 45 ).Laut Lorenz et al. ist der wichtigste Faktor für die Entwicklung einer CTEPH das Vorliegen eines vollständigen Verschlusses der zentralen Lungenarterien, der Lungenlappenarterien oder beider bei der ersten Bildgebung. Ein vergrößerter PA-Durchmesser und das Vorliegen einer Mosaikabschwächung können ebenfalls weitere Prädiktoren sein ( 46 ).Zu den beitragenden Faktoren zählen Gerinnungs- und Fibrinolyseanomalien, beeinträchtigte Thrombozytenfunktion, Gefäßumbau, Entzündungen, Blutgruppen und Krebs ( 45 ). Im Gegensatz zu klassischen genetischen Thromboserisikofaktoren werden Lupus-Antikoagulans- (LAC) und Anti-Phospholipid- (APL) Antikörper häufiger mit CTEPH in Verbindung gebracht ( 6 ). Fibrinogenpolymorphismus und Faktor-V-Leiden-Mutationen tragen zur Resistenz von Thromben gegen Plasminaktivität bei ( 53 ), während sich Thrombozytenerkrankungen als erhöhter Umsatz und beeinträchtigte Aggregation manifestieren ( 54 , 55 ).Entzündungen spielen eine bedeutende Rolle. Erhöhte Werte des C-reaktiven Proteins (CRP) und anderer Entzündungsmarker sind bei CTEPH-Patienten deutlich häufiger ( 56 ). Zu den Risikofaktoren zählen außerdem Krebserkrankungen, Schilddrüsenersatztherapie, andere Blutgruppen als 0, ventrikuloatriale Shunts, infizierte Herzschrittmacher, Splenektomie und frühere rezidivierende venöse Thromboembolien (VTE) ( 57 ). Obwohl unser Verständnis der CTEPH-Pathophysiologie fortgeschritten ist, müssen viele Aspekte, darunter die Rolle von BMPR2-Mutationen und microRNAs, noch vollständig geklärt werden ( 58 ).
5 Molekulare Mechanismen der Mikrovaskulopathie bei CTEPHCTEPH ist eine vielschichtige Erkrankung mit einem komplexen molekularen Hintergrund. Polymorphismen wie jene in den Genen ENG und MAPK10, Mutationen in BMPR2 und SMAD9 sowie dysregulierte Genexpressionen, wie jene, die den PPAR-Signalweg betreffen, sind mit der Pathophysiologie der Erkrankung in Zusammenhang gebracht worden. Die Veränderung in ENG führt zu einer Resistenz gegen Fibrinolyse, während MAPK10-Veränderungen den MAPK-Signalweg beeinflussen, der für die Zellkommunikation und -proliferation entscheidend ist ( 60 ). Mutationen in BMPR2 stören die TGF-β-Signalgebung und induzieren die Proliferation von pulmonalarteriellen glatten Muskelzellen (PASMC), was auf eine zentrale Rolle bei der Gefäßumgestaltung hindeutet, einem Kennzeichen von CTEPH ( 61 , 62 ).Ein tieferer Einblick in die Genexpression offenbart ein dysreguliertes Orchester aus Chemokinsignalen, Stoffwechselwegen und Arachidonsäurestoffwechsel, was auf Entzündungen und Stoffwechselverschiebungen im Kern der Krankheit hinweist. Die identifizierten Mikro-RNAs und DNA-Methylierungsmuster bei Patienten mit CTEPH liefern weitere Einblicke in die komplexen Regulationsmechanismen, die hier eine Rolle spielen und Prozesse vom Fibrinogenabbau bis zur TGF-β-Signalisierung und PASMC-Migration beeinflussen. Insbesondere die Herunterregulierung von miR-759 und Veränderungen in der let-7d-5p-miR-204-5p-Achse deuten auf neue Bereiche für therapeutisches Targeting und prognostische Bewertung hin ( 61 ).Jüngste Studien haben die bedeutende Rolle des NLRP3-Inflammasoms bei PH hervorgehoben und es als potenzielles therapeutisches Ziel für CTEPH vorgeschlagen. Die Aktivierung des NLRP3-Inflammasoms führt zu einem entzündungsfördernden programmierten Zelltod, der als Pyroptose bezeichnet wird, durch die autoproteolytische Aktivierung von Caspase-1, die anschließend die Spaltung der entzündungsfördernden Zytokine IL-1β und IL-18 auslöst ( 63 , 64 ). Diese Entzündungskaskade kann zu der für CTEPH charakteristischen Umgestaltung der Lungengefäße beitragen, was die Aktivierung des Inflammasoms als kritischen Bereich für eine therapeutische Intervention kennzeichnet ( 65 ). Interessanterweise haben Naturprodukte und -formulierungen, einschließlich Curcumin, Resveratrol, Triptolid und Allicin, eine schützende Wirkung gegen hypertensive Organschäden durch Hemmung des NLRP3-Inflammasoms gezeigt ( 66 ).Die Integration dieser molekularen Erkenntnisse ist für die Gestaltung präklinischer Studien von entscheidender Bedeutung und könnte möglicherweise zu neuen therapeutischen Strategien führen, die auf diese spezifischen Signalwege und Mechanismen abzielen. Dieser Bericht bietet zwar einen umfassenden Überblick über die molekulare Landschaft von CTEPH, doch ist kontinuierliche Forschung auf diesem sich rasch entwickelnden Gebiet von entscheidender Bedeutung, um diese Ziele zu validieren und ihre Rolle bei Krankheitsverlauf und -behandlung zu verstehen ( 60 ).Bei Patienten mit CTEPH, die entweder inoperabel sind oder nach PEA weiterhin an PH leiden, konnten durch die Gabe von Riociguat klinische Fortschritte beobachtet werden. Dieses Medikament, ein Stimulator der löslichen Guanylatcyclase (sGC), unterstreicht die entscheidende Rolle des Stickoxid (NO)-sGC-cGMP-Signalwegs bei der Behandlung der Erkrankung ( 49 ). Diese spielen eine zentrale Rolle als Vasodilatatoren und hemmen gleichzeitig die Leukozytenadhäsion, die Thrombozytenaggregation sowie die Proliferation und Migration von Gefäßglattmuskelzellen. Passend dazu hat Macitentan, ein Endothelin-1 (ET-1)-Rezeptorantagonist, in klinischen Studien an Patienten mit inoperabler CTEPH vielversprechende Ergebnisse gezeigt, was das Potenzial gezielter Angriffe auf die NO-, ET-1- und Prostacyclin (PGI2)-Signalwege in Behandlungsstrategien unterstreicht, die denen bei PAH ähneln ( 49 , 67 ) ( Abbildung 4 ).Bei CTEPH kommt es zu erheblichen Veränderungen des rechten Ventrikels und der Lungenzirkulation ( 58 ). Das Konzept der ventrikuloarteriellen Kopplung ist von zentraler Bedeutung, da es die optimale Anpassung der Funktion des rechten Ventrikels an die Lungengefäßbelastung für eine effiziente Energieübertragung widerspiegelt ( 58 , 59 ). Gut angepasste rechte Ventrikel halten eine effiziente ventrikuloarterielle Kopplung aufrecht, während fehlangepasste Ventrikel eine gestörte Kopplung aufweisen, was die Effizienz der Energieübertragung beeinträchtigt ( 58 , 59 ). Trotz reduzierter rechtsventrikulärer Ejektionsfraktion (RVEF) bei CTEPH nimmt die ventrikuläre Elastizität häufig zu, was auf eine adaptive Reaktion auf einen erhöhten PVR hindeutet ( 59 ).Aufgrund ihrer anatomischen Verbindung sind beide Ventrikel betroffen. Die gegenseitige Abhängigkeit der Ventrikel wird durch eine abnorme Septumbewegung während der frühen Diastole des linken Ventrikels deutlich ( 58 ). Diese Bewegung resultiert aus einer dyssynchronen Entspannung zwischen den Ventrikeln und weist auf eine Überlastung des rechten Ventrikels bei CTEPH hin ( 58 ). Die nach links gerichtete Septumbewegung während der frühen Diastole aufgrund der späten rechtsventrikulären Auswurfleistung trägt zur Unterfüllung und Atrophie des linken Ventrikels bei ( 58 , 59 ).Bei schwerer CTEPH wird eine erhöhte diastolische Steifheit des rechten Ventrikels beobachtet, die manchmal von Fibrose und biologischen Veränderungen wie verminderter Titinphosphorylierung begleitet wird ( 58 ). Die komplexe Beziehung zwischen dem rechten Ventrikel, der arteriellen Belastung und der interventrikulären Interaktion ist von entscheidender Bedeutung für das Verständnis der Pathophysiologie von CTEPH, da sie die adaptiven und maladaptiven Veränderungen im rechten Ventrikel und seine Kopplung mit dem Lungenkreislauf hervorhebt ( Abbildungen 2 , 3 ). Abildung 2
Chronische thromboembolische pulmonale Hypertonie (CTEPH) stellt aufgrund ihrer komplexen und häufig unspezifischen klinischen Erscheinungsformen eine erhebliche diagnostische Herausforderung dar. Dieser Bericht skizziert einen umfassenden Ansatz zur diagnostischen Beurteilung von CTEPH und betont, wie wichtig ein hoher Verdachtsindex bei Patienten mit unerklärlicher Dyspnoe oder anhaltenden Symptomen nach akuter Lungenembolie ist. Wir erörtern die zentrale Rolle multimodaler Bildgebung, darunter Echokardiografie, Ventilations-/Perfusionsscans, CT-Pulmonalisangiografie und Magnetresonanztomographie bei der Identifizierung und Bestätigung von CTEPH. Außerdem unterstreicht der Bericht die wesentliche Funktion der Rechtsherzkatheterisierung bei der Validierung der hämodynamischen Parameter, die auf CTEPH hinweisen, und der Sicherstellung der definitiven Diagnose. Fortschritte bei Diagnosetechnologien und die Integration eines multidisziplinären Ansatzes sind für eine rechtzeitige und genaue Diagnose von CTEPH von entscheidender Bedeutung, da sie frühe therapeutische Eingriffe ermöglichen und die Patientenergebnisse verbessern. Ziel dieses Manuskripts ist es, Klinikärzte mit dem Wissen und den Werkzeugen auszustatten, die für einen effizienten Diagnoseablauf bei CTEPH erforderlich sind, und das Bewusstsein und Verständnis für diese potenziell behandelbare Ursache der pulmonalen Hypertonie zu fördern.1 EinleitungPulmonale Hypertonie (PH) ist definiert durch einen erhöhten mittleren pulmonalarteriellen Druck (mPAP), gemessen durch eine Rechtsherzkatheterisierung (RHC) und kann durch verschiedene Erkrankungen des Lungenkreislaufs verursacht werden ( 1 ). Sie ist oft mit Herz-Kreislauf- und Atemwegserkrankungen verbunden und wird durch Zellproliferation und Fibrose in den kleinen Lungenarterien verursacht, was zu einem erhöhten pulmonalvaskulären Widerstand (PVR) und schließlich zu einer Rechtsherzinsuffizienz führt, die die Hauptursache für Morbidität und Mortalität bei PAH ist ( 2 ). Die Behandlung von PH erfordert einen multidisziplinären Ansatz, bei dem die Zusammenarbeit zwischen Patienten und medizinischem Personal im Vordergrund steht ( 3 ).Leichte Erhöhungen des pulmonalarteriellen Drucks sind signifikante Prädiktoren der Mortalität und unterstreichen die Notwendigkeit einer frühzeitigen Erkennung und Behandlung der PH ( 4 ).Chronische thromboembolische pulmonale Hypertonie (CTEPH) ist eine schwere Form der PH, die durch intraluminale Thrombosen, fibröse Gewebe und Obliteration der Pulmonalarterien gekennzeichnet ist ( 5 ). Die genauen molekularen Auslöser für die umfassenden Umbauvorgänge und die Fibrose bei CTEPH sind unbekannt. Man geht davon aus, dass die Erkrankung durch nicht aufgelöstes thromboembolisches Material in den Pulmonalarterien verursacht wird, wo ein niedriger Sauerstoffpartialdruck die Transkription von Genen fördert, die an Gerinnung, Angiogenese, Entzündung und Fibrose beteiligt sind. Diese Erkrankung ist durch ein proliferatives und entzündliches Zellumfeld gekennzeichnet, das zu Fibrose, Narbenbildung und okklusiven Läsionen führt ( 6 ). Bei der Behandlung dieser Patienten ist eine Risikostratifizierung von entscheidender Bedeutung ( 7 ).In diesem Bericht werden die grundlegenden Mechanismen von pulmonal-hypertensiven Erkrankungen erörtert, auf den aktuellen Kenntnisstand zur Pathobiologie von CTEPH eingegangen und Diagnoseinstrumente sowie Behandlungsstrategien zur Erzielung optimaler Behandlungsergebnisse für die Patienten dargelegt. 2 Definitionen und KlassifikationenIm Jahr 2009 analysierten Kovacs et al. RHC-Daten von gesunden Personen, um Normalwerte für den mittleren pulmonalarteriellen Druck (mPAP) in Ruhe und unter Belastung festzulegen. Sie überprüften Daten von 1.187 gesunden Personen aus 47 Studien und fanden einen Ruhe-mPAP von 14,0 ± 3,3 mmHg, ein Wert, der für alle Geschlechter und Ethnien gleich ist und nur minimalen Einfluss von Alter und Körperhaltung hat. Ein Ruhe-mPAP von über 20 mmHg – abgeleitet durch die Addition zweier Standardabweichungen – wurde als Überschreitung der normalen Obergrenze (über dem 97,5. Perzentil) identifiziert und korrelierte mit einem erhöhten Mortalitätsrisiko aller Ursachen. Der durch Belastung bedingte mPAP liegt oft über 30 mmHg, insbesondere bei älteren Erwachsenen, was die Festlegung von Standardwerten für den mPAP unter Belastung erschwert ( 3 , 4 , 8 , 9 ). Die ERS Task Force definiert PH unter Belastung vorläufig als einen mPAP von über 30 mmHg bei einem totalen Lungenwiderstand (TPR) von über 3 Wood-Einheiten (WU) bei maximaler Belastung, wobei alternativ eine mPAP/Herzleistung (CO)-Steigung von über 3 WU als Grenzwert vorgeschlagen wird. Die Steigungen von mPAP/CO und pulmonalarteriellem Verschlussdruck (PAWP)/CO sind entscheidend für die Beurteilung der Lungenzirkulation unter Belastung, da sie stark altersabhängig sind und bei unterschiedlichen Belastungen gleichbleibend sind. Eine PAWP/CO-Steigung von über 2 WU wird mit negativen kardiovaskulären Folgen in Verbindung gebracht und hilft bei der Unterscheidung zwischen prä- und postkapillären Ursachen von PH unter Belastung ( 10 ). Erhöhte mPAP-Werte unter bestimmten physiologischen Zuständen sind allein kein Hinweis auf eine pulmonalarterielle Gefäßerkrankung. Der PVR ist für die Prognose und die klinische Entscheidungsfindung bei pulmonaler arterieller Hypertonie (PAH) von entscheidender Bedeutung. Die vorliegenden Daten deuten darauf hin, dass die obere Normgrenze und der niedrigste prognostisch relevante PVR-Schwellenwert bei etwa 2 WU liegen ( 11 – 13 ).Die ESC/ERS-Leitlinien 2022 zur Diagnose und Behandlung von pulmonaler Hypertonie klassifizieren PH basierend auf den mPAP-, PAWP- und PVR-Werten in drei Haupttypen:• Präkapilläre pulmonale Hypertonie: mPAP >20 mmHg, PAWP <15 mmHg, PVR >2 WU.• Postkapilläre pulmonale Hypertonie (IpcPH): mPAP >20 mmHg, PAWP >15 mmHg, PVR <2 WU.• Kombinierte pulmonale Hypertonie (CpcPH): mPAP >20 mmHg, PAWP >15 mmHg, PVR >2 WU. Darüber hinaus sind die Belastungs-PH und die nicht klassifizierte PH anerkannt. Zu letzterer zählen Patienten mit mPAP >20 mmHg, PVR <2 WU und PAWP <15 mmHg, oft in Verbindung mit einer erhöhten Lungendurchblutung und Erkrankungen wie angeborenen Herzfehlern, Lebererkrankungen oder Lungenerkrankungen, was weitere ätiologische Studien und klinische Nachuntersuchungen erforderlich macht ( 3 ).PH wird anhand pathophysiologischer, klinischer und therapeutischer Aspekte in fünf Gruppen eingeteilt: • Gruppe 1: Pulmonale arterielle Hypertonie (PAH) • Gruppe 2: PH aufgrund einer linksseitigen Herzerkrankung • Gruppe 3: PH aufgrund von Lungenerkrankungen oder Hypoxie • Gruppe 4: PH in Verbindung mit einer Obstruktion der Lungenarterien (kann auf CTEPH, thrombotische Mikroangiopathie durch Lungentumoren oder andere Ursachen für eine Obstruktion der Lungenarterien zurückzuführen sein, beispielsweise Sarkome (hoch- oder mittelgradig oder Angiosarkom), andere bösartige Tumoren (z. B. Nierenkarzinom, Uteruskarzinom, Keimzelltumoren der Hoden) oder nicht bösartige Tumoren (z. B. Uterusleiomyom), Arteriitis ohne Bindegewebserkrankung, angeborene Lungenarterienstenosen und Echinokokkose) ( 14 ) • Gruppe 5: PH mit unklaren oder multifaktoriellen Mechanismen Unabhängig von der zugrundeliegenden Ursache ist die Entwicklung einer PH mit einer Verschlechterung der Symptome und einem erhöhten Sterberisiko verbunden ( 14 ).Zur pulmonal-arteriellen Hypertonie (PAH) der Gruppe 1 gehören: • Idiopathische PAH • Erbliche PAH • Arzneimittel- und toxinbedingte PAH • PAH verbunden mit:○ Bindegewebserkrankungen○ HIV-Infektion○ Portale Hypertonie○ Angeborene Herzerkrankungen○ Bilharziose○ Chronische hämolytische Anämie Diese Klassifikation orientiert sich an den Leitlinien der ESC/ERS und gewährleistet eine umfassende Abdeckung der verschiedenen Ursachen der PAH ( Ergänzende Tabelle 1 ).Gruppe 2 umfasst die postkapilläre PH, entweder isoliert oder kombiniert mit einer präkapillären Komponente. Sie tritt häufig bei Herzinsuffizienz mit erhaltener oder reduzierter Ejektionsfraktion auf und betrifft über 50 % der entsprechenden Patienten ( 15 , 16 ). In beiden Patientengruppen ist das Mortalitätsrisiko bei Funktionsstörungen des rechten Ventrikels doppelt so hoch. Ebenso entwickeln 50–70 % der Patienten mit schwerer Aortenstenose eine PH, wodurch sich ihr Mortalitätsrisiko verdoppelt ( 17 , 18 ). Ebenso wird eine PH bei 60–70 % der Patienten mit schwerer, symptomatischer Mitralklappenerkrankung beobachtet, wobei die Prävalenz mit zunehmender Schwere der linksseitigen Klappenerkrankungen zunimmt ( 19 ).Patienten mit Herzinsuffizienz und erhaltener Ejektionsfraktion (HFpEF), die in erster Linie an Belastungsdyspnoe leiden, können in Ruhe normale Füllungsdrücke, aber bei leichter Belastung deutlich erhöhte Drücke aufweisen ( 20 ). Diese Patientengruppe, bei der es sich oft um ältere Menschen handelt, zeigt während der Belastung eine Abnahme des Herzzeitvolumens aufgrund einer verringerten ventrikulären Compliance und Relaxation mit dem Alter. Belastungstests können frühe Anzeichen einer HFpEF aufdecken, selbst bei normalen Ruhewerten des pulmonalkapillären Verschlussdrucks (PCWP) (<15 mmHg). Bemerkenswerterweise weisen 30 % der gesunden Personen über 60 Jahre bei Belastungstests PCWP-Werte über 25 mmHg auf, was auf den potenziellen Nutzen altersangepasster PCWP-Diagnoseschwellenwerte für eine verbesserte Diagnosegenauigkeit hindeutet ( 21 ).Bei Patienten mit isolierter postkapillärer PH aufgrund einer Gefäßstauung infolge einer Linksherzerkrankung können frühe Interventionen wie die Stabilisierung des intravaskulären Volumens oder die Behebung des primären Defekts die PH oft rückgängig machen, bevor es zu signifikanten Gefäßumbauvorgängen und einem erhöhten PVR kommt ( 22 ). Dies unterstreicht die dringende Notwendigkeit von Strategien, die auf eine frühe Erkennung und Diagnose der PH abzielen, um irreversible Gefäßveränderungen, die zu erhöhtem mPAP führen, zu verhindern.In Gruppe 3 ist die PH typischerweise leicht ausgeprägt, beeinträchtigt die Patienten jedoch erheblich, indem sie die Symptome verschlimmern, die körperliche Belastbarkeit verringern, die Zahl der Krankenhauseinweisungen erhöhen und die Sterblichkeit im Vergleich zu Patienten ohne PH erhöhen ( 14 ).Gruppe 4 konzentriert sich auf CTEPH, eine Erkrankung, die durch Symptome gekennzeichnet ist, die durch die Verstopfung der Lungenarterien durch chronische fibrotische Gerinnsel entstehen. Das Fortbestehen dieser Thromben und die daraus resultierende mikrovaskuläre Arteriopathie erhöhen den pulmonalen Gefäßwiderstand, was zu PH und Rechtsherzversagen führt. Einzigartig ist, dass CTEPH potenziell durch eine Lungenendarteriektomie (PEA) heilbar ist. Für die Diagnose ist eine Rechtsherzkatheteruntersuchung erforderlich, um Folgendes zu bestätigen:1. mPAP > 20 mmHg, PAWP ≤ 15 mmHg in Ruhe und PVR > 2 WU;2. Positive Befunde in der Lungenbildgebung, wie etwa Nichtübereinstimmungen bei der Ventilations-/Perfusionsszintigraphie (V/Q), erhebliche Perfusionsdefekte bei der Einzelphotonen-Emissionscomputertomographie (SPECT), Lungengefäßanomalien bei der CT/MRT oder Lungenangiographie, die auf chronische Verschlüsse hinweisen;3. Bestätigung dieser Zustände nach mindestens drei Monaten unter effektiver Antikoagulation, um eine Unterscheidung von einer subakuten Lungenembolie zu ermöglichen ( 3 , 23 – 26 ).
CTEPH und Chronische Thromboembolische Erkrankung (CTED) sind Begriffe, die symptomatische Personen mit chronischen Pulmonalarterienverschlüssen anhand des Vorhandenseins oder Fehlens von PH in Ruhe unterscheiden ( 24 ). CTED beschreibt Patienten mit mPAP <25 mmHg, anhaltenden Gefäßverschlüssen, Belastungsintoleranz und erheblichen Auswirkungen auf Symptom und Lebensqualität, denen eine PH in Ruhe fehlt. Trotz ähnlicher Symptome wie bei CTEPH können CTED-Patienten ähnliche chirurgische oder interventionelle Behandlungen erhalten. Ihre Belastungseinschränkung rührt von der belastungsbedingten PH und der Totraumventilation her ( 27 ). Die neuen mPAP- und PVR-Kriterien der Leitlinien von 2022 haben zu einer Neuklassifizierung einiger CTED-Patienten als CTEPH geführt und den Begriff „chronische thromboembolische Lungenerkrankung“ (CTEPD) eingeführt, um symptomatische Patienten mit Durchblutungsstörungen und chronischen Gerinnselzeichen unabhängig von der Ruhe-PH zu umfassen ( 24 ). Der Übergang von CTEPD-Patienten zu CTEPH und die Faktoren, die ihre Prognose beeinflussen, bleiben weiterhin Gegenstand eingehender Untersuchungen ( 25 , 28 ).Zur Gruppe 5 zählen Erkrankungen, die aufgrund vielfältiger und komplexer Mechanismen häufig mit PH in Zusammenhang stehen; hierzu zählen etwa chronisches Nierenversagen und Sarkoidose, die durch unterschiedliche pathophysiologische Ursachen charakterisiert sind ( 14 ).Bei belastungsbedingter PAH sind die mPAP/CO- und PAWP/CO-Steigungen entscheidend, da sie unabhängig von der Belastung Erkenntnisse liefern, aber stark altersabhängig sind. Eine mPAP/CO-Steigung von >3 mmHg/l/min zwischen Ruhe und Belastung ist für die Diagnose unverzichtbar, da höhere Steigungen auf eine schlechtere Überlebenschance bei allen kardiopulmonalen Erkrankungen hinweisen. Eine PAWP/CO-Steigung von >2 WU ist mit kardiovaskulären Risiken verbunden und hilft bei der Unterscheidung zwischen prä- und postkapillären PH-Ursachen bei körperlicher Belastung ( 10 ) ( Abbildung 1 ).ng 13 Epidemiologie und RisikofaktorenDie Prävalenz der PAH, kategorisiert als PH der Gruppe 1, beträgt etwa 25 Fälle pro 1 Million Einwohner, mit einer jährlichen Inzidenz von etwa fünf Fällen pro 1 Million Einwohner ( 29 , 30 ). Das mediane Alter bei Diagnose in klinischen Studien und Registern beträgt etwa 53 Jahre ( 31 ).Studien für Gruppe 4 zeigen, dass 1,0–8,8 % der Patienten, die eine akute Lungenembolie (PE) überleben, an CTEPH erkranken können, wobei die Inzidenz laut Chausheva et al. ( 5 ) bei etwa 3 % liegt. Die von Registern in Spanien und Großbritannien gemeldeten jährlichen Inzidenzraten betragen 0,9 bzw. 1,75 pro Million ( 32 , 33 ). Deutsche Daten deuten auf eine Inzidenz von 4,0 pro Million Erwachsene pro Jahr hin ( 14 ), die im Jahr 2016 auf 5,7 anstieg, während Frankreich 5–6 Fälle pro Million Erwachsene meldet ( 34 ). Die Registerdaten variieren und deuten auf eine CTEPH-Inzidenz und -Prävalenz von 2–6 bzw. 26–38 Fällen pro Million Erwachsene hin ( 35 – 37 ). Die steigende Zahl diagnostizierter CTEPH-Fälle ist auf ein besseres Verständnis der Krankheit und aktives Screening zurückzuführen, insbesondere bei Patienten, die nach einer PE symptomatisch bleiben oder ein Risiko für CTEPH haben ( 24 , 34 , 38 ). Die weltweite Prävalenz und Inzidenz von CTEPH sind weiterhin unklar, so dass für genaue Definitionen weitere Forschung erforderlich ist ( Abbildung 1 ) ( 28 ).In Europa und den USA beträgt die durchschnittliche Inzidenz von CTEPH etwa 4 %, im Gegensatz zu bis zu 14 % in Japan ( 15 , 39 ), wo die Lungenembolierate wesentlich niedriger ist. In Europa sind beide Geschlechter gleichermaßen von der Krankheit betroffen, während in Japan Frauen überwiegen ( 5 ). Das europäische CTEPH-Register zeigt, dass bei Frauen bestimmte kardiovaskuläre Risikofaktoren weniger häufig auftreten, jedoch Übergewicht, Krebs und Schilddrüsenerkrankungen häufiger auftreten ( 26 ). Bemerkenswerterweise haben 50–75 % der CTEPH-Patienten eine dokumentierte Vorgeschichte einer akuten Lungenembolie, während dieser Wert bei japanischen Patienten auf 15 % sinkt, was auf einen bestimmten CTEPH-Phänotyp hindeutet ( 24 ).Alle Metaanalysen berichten von einer CTEPH-Inzidenz von 2,7–3 %, wobei etwa 30 % der Patienten eine CTED entwickeln, eine symptomatische Restvaskulopathie ohne PH ( 23 , 34 , 35 ). In der FOCUS-Studie wurde insbesondere eine Inzidenz von Beeinträchtigungen nach einer Lungenembolie von 16 % berichtet, wobei CTEPH bei 2,3 % der Patienten auftrat ( 40 ). Es wurde auch berichtet, dass bis zu 20 % der Patienten nach einer Lungenembolie irgendeine Form von Beeinträchtigung erfahren können, was die Notwendigkeit einer aufmerksamen Überwachung und eines aktiven Screenings bei diesen Patienten unterstreicht, um langfristige Komplikationen wirksam behandeln und eindämmen zu können ( 40 ).Zu den Risikofaktoren für CTEPH zählen venöse Thromboembolien (VTE) in der Vorgeschichte, insbesondere wiederkehrende VTE, erhöhter pulmonalarterieller Druck nach Lungenembolie, maligne Erkrankungen in der Vorgeschichte, Splenektomie, Staphylokokkeninfektion, Blutgruppen außer O, Antiphospholipid-Antikörper, Lupusantikoagulans und permanente intravaskuläre Geräte. Weitere Risikofaktoren sind entzündliche Darmerkrankungen, Polycythaemia vera, essentielle Thrombozythämie, Hypothyreose, erhöhte Werte von Faktor VIII und von-Willebrand-Faktor, Fibrinogen-Polymorphismus und möglicherweise Faktor V Leiden, obwohl die Rolle des Letzteren noch umstritten ist ( 3 , 24 , 40 – 42 ). Demografische Merkmale und das Vorhandensein von chronischen entzündlichen/Autoimmunerkrankungen oder hämatologischen Erkrankungen spielen ebenfalls eine wichtige Rolle bei der Entstehung der Erkrankung ( 43 , 44 ) ( Tabelle 1 ).
CTEPH ist gekennzeichnet durch einen prothrombotischen Zustand und abnorme Fibrinogenmoleküle, die einer physiologischen Thrombolyse widerstehen, was auf eine beeinträchtigte Auflösung des anfänglichen embolischen Ereignisses hinweist. Eine Splenektomie ist insbesondere mit distalen CTEPH-Typen assoziiert und erschwert PEA-Verfahren ( 44 ). Eine Vorgeschichte infizierter ventrikuloatrialer Shunts und Splenektomien ist nicht nur mit CTEPH, sondern auch mit erheblichen Entzündungen und einem erhöhten VTE-Risiko verbunden ( 41 ). Darüber hinaus kann CTEPH durch eine postkapilläre Krankheitskomponente kompliziert werden, was die Bedeutung der Erkennung klinischer Risikofaktoren für PH im Zusammenhang mit Linksherzerkrankungen unterstreicht ( 47 ).4 PathophysiologieCTEPH ist durch vaskuläre Umbauprozesse in der Lunge gekennzeichnet, die sowohl verschlossene als auch nicht verschlossene kleine Gefäße betreffen, was zu hohen Morbiditäts- und Mortalitätsraten führt, insbesondere aufgrund der Progression zu einer Rechtsherzinsuffizienz, wenn die Erkrankung unbehandelt bleibt ( 24 , 48 , 49 ). CTEPH ist eine seltene Spätkomplikation einer APE, die auch nach mehr als drei Monaten Antikoagulationstherapie nicht abklingt und typischerweise auf eine venöse Thromboembolie zurückzuführen ist ( 50 ). Etwa 4 % der APE-Fälle entwickeln sich innerhalb von 2 Jahren zu CTEPH ( 51 ), wobei 75 % der CTEPH-Patienten eine APE-Anamnese haben ( 52 ).Der Grund, warum manche Menschen nach einer APE eine CTEPH entwickeln, ist weiterhin unklar, insbesondere angesichts der Tatsache, dass sich die meisten Gerinnsel unter Antikoagulation auflösen. CTEPH-Thromben sind faserig und weisen kollagene und entzündliche Komponenten auf, was manchmal zur Verkalkung führt ( 45 ).Laut Lorenz et al. ist der wichtigste Faktor für die Entwicklung einer CTEPH das Vorliegen eines vollständigen Verschlusses der zentralen Lungenarterien, der Lungenlappenarterien oder beider bei der ersten Bildgebung. Ein vergrößerter PA-Durchmesser und das Vorliegen einer Mosaikabschwächung können ebenfalls weitere Prädiktoren sein ( 46 ).Zu den beitragenden Faktoren zählen Gerinnungs- und Fibrinolyseanomalien, beeinträchtigte Thrombozytenfunktion, Gefäßumbau, Entzündungen, Blutgruppen und Krebs ( 45 ). Im Gegensatz zu klassischen genetischen Thromboserisikofaktoren werden Lupus-Antikoagulans- (LAC) und Anti-Phospholipid- (APL) Antikörper häufiger mit CTEPH in Verbindung gebracht ( 6 ). Fibrinogenpolymorphismus und Faktor-V-Leiden-Mutationen tragen zur Resistenz von Thromben gegen Plasminaktivität bei ( 53 ), während sich Thrombozytenerkrankungen als erhöhter Umsatz und beeinträchtigte Aggregation manifestieren ( 54 , 55 ).Entzündungen spielen eine bedeutende Rolle. Erhöhte Werte des C-reaktiven Proteins (CRP) und anderer Entzündungsmarker sind bei CTEPH-Patienten deutlich häufiger ( 56 ). Zu den Risikofaktoren zählen außerdem Krebserkrankungen, Schilddrüsenersatztherapie, andere Blutgruppen als 0, ventrikuloatriale Shunts, infizierte Herzschrittmacher, Splenektomie und frühere rezidivierende venöse Thromboembolien (VTE) ( 57 ). Obwohl unser Verständnis der CTEPH-Pathophysiologie fortgeschritten ist, müssen viele Aspekte, darunter die Rolle von BMPR2-Mutationen und microRNAs, noch vollständig geklärt werden ( 58 ).
5 Molekulare Mechanismen der Mikrovaskulopathie bei CTEPHCTEPH ist eine vielschichtige Erkrankung mit einem komplexen molekularen Hintergrund. Polymorphismen wie jene in den Genen ENG und MAPK10, Mutationen in BMPR2 und SMAD9 sowie dysregulierte Genexpressionen, wie jene, die den PPAR-Signalweg betreffen, sind mit der Pathophysiologie der Erkrankung in Zusammenhang gebracht worden. Die Veränderung in ENG führt zu einer Resistenz gegen Fibrinolyse, während MAPK10-Veränderungen den MAPK-Signalweg beeinflussen, der für die Zellkommunikation und -proliferation entscheidend ist ( 60 ). Mutationen in BMPR2 stören die TGF-β-Signalgebung und induzieren die Proliferation von pulmonalarteriellen glatten Muskelzellen (PASMC), was auf eine zentrale Rolle bei der Gefäßumgestaltung hindeutet, einem Kennzeichen von CTEPH ( 61 , 62 ).Ein tieferer Einblick in die Genexpression offenbart ein dysreguliertes Orchester aus Chemokinsignalen, Stoffwechselwegen und Arachidonsäurestoffwechsel, was auf Entzündungen und Stoffwechselverschiebungen im Kern der Krankheit hinweist. Die identifizierten Mikro-RNAs und DNA-Methylierungsmuster bei Patienten mit CTEPH liefern weitere Einblicke in die komplexen Regulationsmechanismen, die hier eine Rolle spielen und Prozesse vom Fibrinogenabbau bis zur TGF-β-Signalisierung und PASMC-Migration beeinflussen. Insbesondere die Herunterregulierung von miR-759 und Veränderungen in der let-7d-5p-miR-204-5p-Achse deuten auf neue Bereiche für therapeutisches Targeting und prognostische Bewertung hin ( 61 ).Jüngste Studien haben die bedeutende Rolle des NLRP3-Inflammasoms bei PH hervorgehoben und es als potenzielles therapeutisches Ziel für CTEPH vorgeschlagen. Die Aktivierung des NLRP3-Inflammasoms führt zu einem entzündungsfördernden programmierten Zelltod, der als Pyroptose bezeichnet wird, durch die autoproteolytische Aktivierung von Caspase-1, die anschließend die Spaltung der entzündungsfördernden Zytokine IL-1β und IL-18 auslöst ( 63 , 64 ). Diese Entzündungskaskade kann zu der für CTEPH charakteristischen Umgestaltung der Lungengefäße beitragen, was die Aktivierung des Inflammasoms als kritischen Bereich für eine therapeutische Intervention kennzeichnet ( 65 ). Interessanterweise haben Naturprodukte und -formulierungen, einschließlich Curcumin, Resveratrol, Triptolid und Allicin, eine schützende Wirkung gegen hypertensive Organschäden durch Hemmung des NLRP3-Inflammasoms gezeigt ( 66 ).Die Integration dieser molekularen Erkenntnisse ist für die Gestaltung präklinischer Studien von entscheidender Bedeutung und könnte möglicherweise zu neuen therapeutischen Strategien führen, die auf diese spezifischen Signalwege und Mechanismen abzielen. Dieser Bericht bietet zwar einen umfassenden Überblick über die molekulare Landschaft von CTEPH, doch ist kontinuierliche Forschung auf diesem sich rasch entwickelnden Gebiet von entscheidender Bedeutung, um diese Ziele zu validieren und ihre Rolle bei Krankheitsverlauf und -behandlung zu verstehen ( 60 ).Bei Patienten mit CTEPH, die entweder inoperabel sind oder nach PEA weiterhin an PH leiden, konnten durch die Gabe von Riociguat klinische Fortschritte beobachtet werden. Dieses Medikament, ein Stimulator der löslichen Guanylatcyclase (sGC), unterstreicht die entscheidende Rolle des Stickoxid (NO)-sGC-cGMP-Signalwegs bei der Behandlung der Erkrankung ( 49 ). Diese spielen eine zentrale Rolle als Vasodilatatoren und hemmen gleichzeitig die Leukozytenadhäsion, die Thrombozytenaggregation sowie die Proliferation und Migration von Gefäßglattmuskelzellen. Passend dazu hat Macitentan, ein Endothelin-1 (ET-1)-Rezeptorantagonist, in klinischen Studien an Patienten mit inoperabler CTEPH vielversprechende Ergebnisse gezeigt, was das Potenzial gezielter Angriffe auf die NO-, ET-1- und Prostacyclin (PGI2)-Signalwege in Behandlungsstrategien unterstreicht, die denen bei PAH ähneln ( 49 , 67 ) ( Abbildung 4 ).Bei CTEPH kommt es zu erheblichen Veränderungen des rechten Ventrikels und der Lungenzirkulation ( 58 ). Das Konzept der ventrikuloarteriellen Kopplung ist von zentraler Bedeutung, da es die optimale Anpassung der Funktion des rechten Ventrikels an die Lungengefäßbelastung für eine effiziente Energieübertragung widerspiegelt ( 58 , 59 ). Gut angepasste rechte Ventrikel halten eine effiziente ventrikuloarterielle Kopplung aufrecht, während fehlangepasste Ventrikel eine gestörte Kopplung aufweisen, was die Effizienz der Energieübertragung beeinträchtigt ( 58 , 59 ). Trotz reduzierter rechtsventrikulärer Ejektionsfraktion (RVEF) bei CTEPH nimmt die ventrikuläre Elastizität häufig zu, was auf eine adaptive Reaktion auf einen erhöhten PVR hindeutet ( 59 ).Aufgrund ihrer anatomischen Verbindung sind beide Ventrikel betroffen. Die gegenseitige Abhängigkeit der Ventrikel wird durch eine abnorme Septumbewegung während der frühen Diastole des linken Ventrikels deutlich ( 58 ). Diese Bewegung resultiert aus einer dyssynchronen Entspannung zwischen den Ventrikeln und weist auf eine Überlastung des rechten Ventrikels bei CTEPH hin ( 58 ). Die nach links gerichtete Septumbewegung während der frühen Diastole aufgrund der späten rechtsventrikulären Auswurfleistung trägt zur Unterfüllung und Atrophie des linken Ventrikels bei ( 58 , 59 ).Bei schwerer CTEPH wird eine erhöhte diastolische Steifheit des rechten Ventrikels beobachtet, die manchmal von Fibrose und biologischen Veränderungen wie verminderter Titinphosphorylierung begleitet wird ( 58 ). Die komplexe Beziehung zwischen dem rechten Ventrikel, der arteriellen Belastung und der interventrikulären Interaktion ist von entscheidender Bedeutung für das Verständnis der Pathophysiologie von CTEPH, da sie die adaptiven und maladaptiven Veränderungen im rechten Ventrikel und seine Kopplung mit dem Lungenkreislauf hervorhebt ( Abbildungen 2 , 3 ). Abildung 2
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.
24 Nov 2024 21:32 #2283
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
danny antwortete auf CTEPH, Hintergründe
6 Klinische MerkmaleDas klinische Erscheinungsbild der CTEPH ist häufig unspezifisch, was eine frühzeitige Diagnose erschwert, da die Symptome subtil sein und leicht mit anderen Erkrankungen verwechselt werden können (
68
). Belastungsdyspnoe oder Kurzatmigkeit bei körperlicher Aktivität ist das am häufigsten von Patienten mit CTEPH berichtete Symptom und beeinträchtigt ihre körperliche Belastbarkeit und ihre Fähigkeit, Aktivitäten, die zuvor Freude bereitet haben, wieder auszuführen, erheblich (
53
).Neben Belastungsdyspnoe können auch andere Symptome auftreten, wenn auch seltener. Patienten können Brustschmerzen verspüren, die von leichtem Unbehagen bis zu starken, stechenden Schmerzen reichen (
49
,
69
). Einige berichten von Herzklopfen, während andere Husten haben können, der in der Regel unproduktiv ist (
49
,
53
). Hämoptyse gilt als relativ selten (
53
).Mit Fortschreiten der CTEPH werden Symptome sichtbar, die auf eine Funktionsstörung des rechten Ventrikels (RV) hinweisen. Ödeme der unteren Extremitäten, die sich als Schwellungen in Beinen und Füßen äußern, sind ein Hinweis auf Flüssigkeitsansammlungen und eine eingeschränkte RV-Funktion (
53
,
58
). Bei körperlicher Anstrengung können Benommenheit, Schwindel und sogar Synkopen (Ohnmacht) auftreten, die auf eine verringerte Herzleistung und unzureichende Durchblutung des Gehirns hinweisen und eine schwerere Phase der Krankheit anzeigen (
53
).Auch die Befunde einer körperlichen Untersuchung können auf CTEPH hinweisen. Eine laute pulmonale Komponente des zweiten Herztons (P2) deutet auf erhöhten pulmonalarteriellen Druck und pulmonalvaskulären Widerstand hin, was auf ein pulmonalarterielles Strömungsgeräusch aufgrund turbulenter Strömung durch partiell verschlossene oder erneut kanülierte chronische Thromben hinweisen kann (
53
). Dieses Anzeichen ist jedoch nicht nur bei CTEPH vorhanden und kann auch bei anderen pulmonalarteriellen Erkrankungen beobachtet werden (
53
,
70
). Das Auftreten eines Trikuspidalklappeninsuffizienzgeräusches weist auf einen Rückfluss des Blutes durch die Trikuspidalklappe hin, oft aufgrund einer Vergrößerung oder Funktionsstörung des rechten Ventrikels (
24
,
53
). In einigen fortgeschrittenen Fällen kann ein tastbarer RV-Hubbel ertastet werden, bei dem der rechte Ventrikel gegen die Brustwand drückt (
53
).Ein weiteres Fortschreiten in Richtung RV-Versagen wird durch zusätzliche Symptome gekennzeichnet. Zunehmende Pulsation der Jugularvene, sichtbar als Ausbeulung der Halsvene, und hepatojugulärer Reflux, bei dem der Druck in der Jugularvene bei Leberkompression steigt, sind auffällig (
24
,
70
). Periphere Ödeme oder Schwellungen in den unteren Extremitäten und anderen abhängigen Bereichen können sich verstärken und insgesamt eine fortschreitende RV-Dysfunktion und den Beginn einer Rechtsherzinsuffizienz signalisieren (
49
,
53
).Im Wesentlichen können die klinischen Merkmale von CTEPH in den verschiedenen Krankheitsstadien erheblich variieren und sind oft unspezifisch, was die Bedeutung eines gründlichen und sorgfältigen Diagnoseprozesses zur Identifizierung dieser potenziell schwerwiegenden Erkrankung unterstreicht.7 DiagnoseIm Laufe der Jahre hat der Ansatz zur Diagnose von PH verschiedene Iterationen und Verbesserungen der Diagnosealgorithmen erfahren. Ein solches innovatives Werkzeug ist das Akronym SCAR – es steht für Suspect, Confirm und Assess Risk (Vermuten, Bestätigen und Risiko einschätzen). Diese Eselsbrücke unterstützt den systematischen Ansatz zur Diagnose von CTEPH und betont die Notwendigkeit eines erhöhten Verdachts, um die Erkrankung genau identifizieren zu können (
71
). Der Nutzen der multimodalen Bildgebung spielt bei der Diagnose von CTEPH eine entscheidende Rolle, was die Komplexität dieser Krankheit und die Notwendigkeit einer umfassenden Bewertung widerspiegelt.7.1 VerdächtigerDer erste Schritt des SCAR-Algorithmus unterstreicht, wie wichtig es ist, einen hohen Verdachtsindex für CTEPH beizubehalten, insbesondere bei Patienten mit unerklärlicher Dyspnoe, einer Lungenembolie in der Anamnese oder Anzeichen einer Rechtsherzinsuffizienz. In dieser Phase geht es darum, das mögliche Vorhandensein von CTEPH anhand der klinischen Präsentation und der Risikofaktoren zu erkennen.7.2 BestätigenBei Verdacht auf CTEPH umfasst die Bestätigung einer CTEPH eine Reihe von diagnostischen Tests, wobei bildgebende Verfahren im Vordergrund stehen. Multimodale Bildgebung, einschließlich Echokardiographie, Ventilations-Perfusions-Scan (V/Q-Scan), Computertomographie-Pulmonalisangiographie (CTPA), Magnetresonanztomographie (MRT) und Lungenangiographie, ist von entscheidender Bedeutung. Jedes dieser Bildgebungsverfahren bietet einzigartige Erkenntnisse: - Die Echokardiographie dient als nicht-invasive Methode zur Beurteilung der Funktion des rechten Ventrikels und zur Schätzung des Lungenarteriendrucks. - Der V/Q-Scan ist für die Erkennung von CTEPH äußerst empfindlich und gilt als Goldstandard-Screening-Test, der Nichtübereinstimmungen aufdecken kann, die auf Lungenembolien hinweisen. - CTPA bietet eine detaillierte Visualisierung der Lungengefäße und ermöglicht so die Identifizierung thromboembolischer Obstruktionen und Gefäßanomalien. - Die MRT bietet zusätzliche funktionelle und strukturelle Informationen über den Herz- und Lungenkreislauf ohne Strahlenbelastung. - Die Pulmonalisangiographie ist nach wie vor die definitive Methode zur Visualisierung der Lungenarterien und bietet genaue Informationen über die Lage und das Ausmaß von Verstopfungen.7.3 RisikobewertungDiese Bewertung ist wichtig, um die Wahrscheinlichkeit der Entwicklung einer CTEPH bei Personen mit einem suggestiven klinischen Muster zu quantifizieren. Nach der Bestätigung der Diagnose umfasst die Risikobewertung die Bewertung des Schweregrads der PH, der Funktion des rechten Ventrikels und des allgemeinen klinischen Zustands des Patienten. Diese Bewertung leitet die Behandlungsstrategie, einschließlich der Entscheidung für einen chirurgischen Eingriff wie PEA oder eine medizinische Behandlung mit zielgerichteten Therapien (
72
).Der SCAR-Algorithmus in Kombination mit multimodaler Bildgebung unterstreicht den personalisierten und schrittweisen Ansatz zur Diagnose von CTEPH. Die Integration fortschrittlicher Bildgebungsverfahren in den Diagnoseprozess hilft nicht nur bei der Bestätigung von CTEPH, sondern liefert auch wichtige Informationen für die individuelle Risikostratifizierung und die anschließende Behandlungsplanung und unterstreicht die wichtige Rolle der Bildgebung bei der umfassenden Betreuung von Patienten mit CTEPH (
Abbildung 5
).
7.4 Blutuntersuchungen und ImmunologieZum Zeitpunkt der Diagnose von CTEPH ist eine umfassende Reihe von Labortests unerlässlich, um den allgemeinen Gesundheitszustand des Patienten zu beurteilen, mögliche Komplikationen zu identifizieren und Behandlungsentscheidungen zu treffen. Die empfohlenen Laboruntersuchungen umfassen eine breite Palette von Tests: - Gesamtblutbild: Unverzichtbar für die Beurteilung des Hämoglobinspiegels, der weißen Blutkörperchen und der Blutplättchen, die auf Anämie, Infektionen oder andere hämatologische Erkrankungen hinweisen können. - Serumelektrolyte: Einschließlich Natrium, Kalium, Magnesium, Chlor, Kalzium und Phosphor, um den Elektrolythaushalt zu beurteilen und mögliche Störungen zu erkennen, die die Herz- und Muskelfunktion beeinträchtigen könnten. - Nierenfunktionstests: Kreatininwerte und die geschätzte glomeruläre Filtrationsrate (eGFR) sind für die Beurteilung der Nierenfunktion von entscheidender Bedeutung, wobei Harnstoffwerte zusätzliche Einblicke in die Nierengesundheit und den Stoffwechselstatus liefern. - Leberfunktionstests: Alanin-Aminotransferase (ALT), Aspartat-Aminotransferase (AST), alkalische Phosphatase, γ-Glutamyl-Transpeptidase (GGT) und Bilirubinwerte helfen bei der Beurteilung der Lebergesundheit und der Erkennung von Leberfunktionsstörungen, die für den Medikamentenstoffwechsel und die allgemeine Gesundheit relevant sein können. - Harnsäure: Erhöhte Werte können auf ein erhöhtes Gichtrisiko hinweisen und mit Herz-Kreislauf-Erkrankungen in Verbindung stehen. - Eisenstatus: Messungen von Serumeisen, Transferrinsättigung und Ferritin sind wichtig für die Erkennung eines Eisenmangels oder einer Eisenüberladung, die Auswirkungen auf Anämie und den allgemeinen Gesundheitszustand haben kann. - BNP oder NT-proBNP: Diese Biomarker der Herzinsuffizienz sind von entscheidender Bedeutung für die Beurteilung der Herzbelastung und die Behandlung von CTEPH. - Serologische Studien: Tests auf Hepatitis- und HIV-Viren sind wichtig, um zugrunde liegende Erkrankungen zu identifizieren, die sich auf Behandlungsmöglichkeiten und Prognose auswirken könnten. - Immunologische Laboruntersuchung: Ein Screening auf antinukleäre Antikörper, Anti-Centromeren-Antikörper und Anti-Ro kann bei der Identifizierung von Autoimmunerkrankungen helfen, die Auswirkungen auf CTEPH haben können. - Biomarker für das Antiphospholipid-Syndrom: Aufgrund der Assoziation des Antiphospholipid-Syndroms mit thrombotischen Ereignissen wird ein Screening empfohlen, obwohl von einem breiteren Thrombophilie-Screening abgeraten wird ( 73 ).7.5 ElektrokardiographieIm klinischen Umfeld ist das 12-Kanal-Elektrokardiogramm (EKG) ein leicht verfügbares Diagnoseinstrument. Zu den EKG-Manifestationen einer Rechtsherzbelastung zählen P-pulmonale (gekennzeichnet durch eine P-Wellen-Amplitude von über 2,5 mm), Rechtsschenkelblock, T-Wellen-Anomalien in den Brustableitungen und eine Rechtsabweichung ( 3 , 74 ). Anzeichen einer Rechtsventrikelhypertrophie (RVH), die auf einen pulmonalarteriellen systolischen Druck (PASP) im Normbereich (<7%) hinweisen, werden selten beobachtet. EKG-Muster mit einem hohen positiven Vorhersagewert (>80%) für pulmonale Hypertonie umfassen R in Ableitung I ≤ 2 mm, S in Ableitung V1 ≤ 2 mm, R/S-Verhältnis in Ableitung V1 ≥ 1, R/S-Verhältnis in Ableitung V6 ≤ 1, QRS-Achse ≥ 110° und ein qR-Muster in V1. Insbesondere eine S-Welle ≤ 2 mm in V1 sagt das Vorhandensein von pulmonaler Hypertonie mit 100-prozentiger Sicherheit voraus. Der Nachweis eines dieser EKG-Muster sollte eine weitere Untersuchung mit transthorakaler Echokardiographie (TTE) zur umfassenderen Beurteilung der Herzfunktion und der PASP-Messung nach sich ziehen. Diese EKG-Muster weisen jedoch niedrige negative Vorhersagewerte auf, was bedeutet, dass ihr Fehlen eine pulmonale Hypertonie nicht endgültig ausschließt ( 75 ). Diese Zusammenfassung umreißt die spezifischen EKG-Kriterien für RVH oder eine Vergrößerung des rechten Vorhofs in Verbindung mit pulmonaler Hypertonie, wie in Abbildung 6 dargestellt .
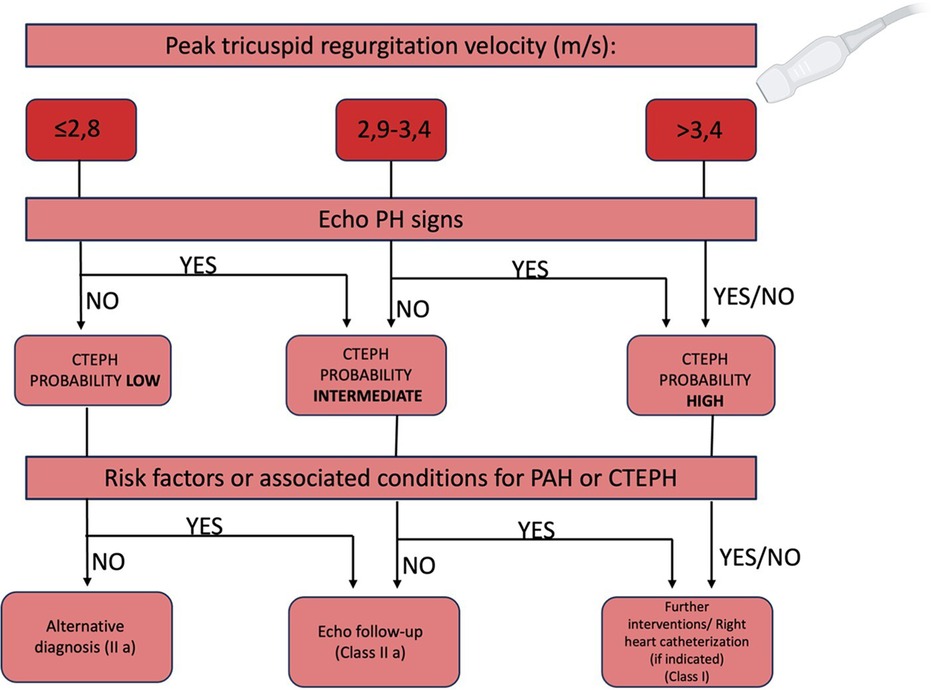
7.6 EchokardiographieDie Leitlinie der Europäischen Gesellschaft für Kardiologie (ESC) zur Lungenembolie (PE) empfiehlt eine Echokardiographie als Erstuntersuchung bei Patienten mit anhaltender Dyspnoe, funktionellen Einschränkungen oder Risikofaktoren für CTEPH ( 3 ). Die TTE ist entscheidend für die Abschätzung des pulmonalarteriellen Drucks und bietet wichtige Einblicke in die Pathophysiologie und Ätiologie von CTEPH und die Auswirkungen der chronischen PAH auf das Herz. Sie liefert auch wichtige Daten zu gleichzeitig bestehenden Herzerkrankungen, Operationsergebnissen und möglichen postoperativen Komplikationen wie Tamponade.Bei Patienten mit bestätigter oder stark vermuteter PH ist eine gründliche Untersuchung unerlässlich, um eine linksseitige Herzerkrankung oder intrakardiale Shunts auszuschließen. Marker wie Dilatation und Dysfunktion des rechten Ventrikels gelten als negative Prognoseindikatoren. Darüber hinaus signalisiert ein Perikarderguss im Rahmen einer PH eine fortgeschrittene Erkrankung mit einer schlechten Prognose ( 76 ).Die echokardiographische Bewertung der PH umfasst die Messung der Spitzengeschwindigkeit der Trikuspidalklappeninsuffizienz, die Berechnung des atrioventrikulären Druckgradienten und die Identifizierung indirekter Zeichen der PH wie Dilatation des rechten Vorhofs und Ventrikels, verminderte Kontraktilität des rechten Ventrikels und Doppler-Flussanomalien im Ausflusstrakt des rechten Ventrikels ( 3 ). Diese Indikatoren müssen jedoch in frühen Krankheitsstadien fehlen, und in 10–31 % der Fälle kann die Echokardiographie die PH übersehen ( 77 ).Der erste Schritt zur echokardiografischen Bestimmung der PH-Wahrscheinlichkeit besteht in der Messung der maximalen Trikuspidalinsuffizienzgeschwindigkeit (TRV). Ein Flussdiagramm zur Ermittlung der CTEPH-Wahrscheinlichkeit empfiehlt die Bewertung der Wahrscheinlichkeit für alle klinischen PH-Gruppen unter Einbeziehung zusätzlicher echokardiografischer Parameter, die in drei Kategorien unterteilt sind, die auf PH hinweisen ( 78 ): - Ventrikel: RV/LV-Basaldurchmesser/Flächenverhältnis >1,0; Abflachung des interventrikulären Septums; TAPSE/sPAP-Verhältnis <0,55 mm/mmHg; - Pulmonalarterie: RVOT-Beschleunigungszeit <105 ms und/oder mittsystolisches Notching; frühdiastolische Pulmonalarteriengeschwindigkeit >2,2 m/s; Durchmesser der Pulmonalarterie größer als Durchmesser der Aortenwurzel oder >25 mm; - Untere Hohlvene und rechter Vorhof: Durchmesser der V. cava >21 mm mit verringertem inspiratorischen Kollaps, Fläche des rechten Vorhofs >18 cm2 . Eine TRV > 3,4 m/s deutet auf eine hohe echokardiografische Wahrscheinlichkeit einer PAH hin, während bei einer TRV ≤ 3,4 m/s zusätzliche Parameter aus mindestens zwei Kategorien erforderlich sind, um die Wahrscheinlichkeit einer PAH zu beurteilen ( Abbildung 7 ). Abildung 7
7.4 Blutuntersuchungen und ImmunologieZum Zeitpunkt der Diagnose von CTEPH ist eine umfassende Reihe von Labortests unerlässlich, um den allgemeinen Gesundheitszustand des Patienten zu beurteilen, mögliche Komplikationen zu identifizieren und Behandlungsentscheidungen zu treffen. Die empfohlenen Laboruntersuchungen umfassen eine breite Palette von Tests: - Gesamtblutbild: Unverzichtbar für die Beurteilung des Hämoglobinspiegels, der weißen Blutkörperchen und der Blutplättchen, die auf Anämie, Infektionen oder andere hämatologische Erkrankungen hinweisen können. - Serumelektrolyte: Einschließlich Natrium, Kalium, Magnesium, Chlor, Kalzium und Phosphor, um den Elektrolythaushalt zu beurteilen und mögliche Störungen zu erkennen, die die Herz- und Muskelfunktion beeinträchtigen könnten. - Nierenfunktionstests: Kreatininwerte und die geschätzte glomeruläre Filtrationsrate (eGFR) sind für die Beurteilung der Nierenfunktion von entscheidender Bedeutung, wobei Harnstoffwerte zusätzliche Einblicke in die Nierengesundheit und den Stoffwechselstatus liefern. - Leberfunktionstests: Alanin-Aminotransferase (ALT), Aspartat-Aminotransferase (AST), alkalische Phosphatase, γ-Glutamyl-Transpeptidase (GGT) und Bilirubinwerte helfen bei der Beurteilung der Lebergesundheit und der Erkennung von Leberfunktionsstörungen, die für den Medikamentenstoffwechsel und die allgemeine Gesundheit relevant sein können. - Harnsäure: Erhöhte Werte können auf ein erhöhtes Gichtrisiko hinweisen und mit Herz-Kreislauf-Erkrankungen in Verbindung stehen. - Eisenstatus: Messungen von Serumeisen, Transferrinsättigung und Ferritin sind wichtig für die Erkennung eines Eisenmangels oder einer Eisenüberladung, die Auswirkungen auf Anämie und den allgemeinen Gesundheitszustand haben kann. - BNP oder NT-proBNP: Diese Biomarker der Herzinsuffizienz sind von entscheidender Bedeutung für die Beurteilung der Herzbelastung und die Behandlung von CTEPH. - Serologische Studien: Tests auf Hepatitis- und HIV-Viren sind wichtig, um zugrunde liegende Erkrankungen zu identifizieren, die sich auf Behandlungsmöglichkeiten und Prognose auswirken könnten. - Immunologische Laboruntersuchung: Ein Screening auf antinukleäre Antikörper, Anti-Centromeren-Antikörper und Anti-Ro kann bei der Identifizierung von Autoimmunerkrankungen helfen, die Auswirkungen auf CTEPH haben können. - Biomarker für das Antiphospholipid-Syndrom: Aufgrund der Assoziation des Antiphospholipid-Syndroms mit thrombotischen Ereignissen wird ein Screening empfohlen, obwohl von einem breiteren Thrombophilie-Screening abgeraten wird ( 73 ).7.5 ElektrokardiographieIm klinischen Umfeld ist das 12-Kanal-Elektrokardiogramm (EKG) ein leicht verfügbares Diagnoseinstrument. Zu den EKG-Manifestationen einer Rechtsherzbelastung zählen P-pulmonale (gekennzeichnet durch eine P-Wellen-Amplitude von über 2,5 mm), Rechtsschenkelblock, T-Wellen-Anomalien in den Brustableitungen und eine Rechtsabweichung ( 3 , 74 ). Anzeichen einer Rechtsventrikelhypertrophie (RVH), die auf einen pulmonalarteriellen systolischen Druck (PASP) im Normbereich (<7%) hinweisen, werden selten beobachtet. EKG-Muster mit einem hohen positiven Vorhersagewert (>80%) für pulmonale Hypertonie umfassen R in Ableitung I ≤ 2 mm, S in Ableitung V1 ≤ 2 mm, R/S-Verhältnis in Ableitung V1 ≥ 1, R/S-Verhältnis in Ableitung V6 ≤ 1, QRS-Achse ≥ 110° und ein qR-Muster in V1. Insbesondere eine S-Welle ≤ 2 mm in V1 sagt das Vorhandensein von pulmonaler Hypertonie mit 100-prozentiger Sicherheit voraus. Der Nachweis eines dieser EKG-Muster sollte eine weitere Untersuchung mit transthorakaler Echokardiographie (TTE) zur umfassenderen Beurteilung der Herzfunktion und der PASP-Messung nach sich ziehen. Diese EKG-Muster weisen jedoch niedrige negative Vorhersagewerte auf, was bedeutet, dass ihr Fehlen eine pulmonale Hypertonie nicht endgültig ausschließt ( 75 ). Diese Zusammenfassung umreißt die spezifischen EKG-Kriterien für RVH oder eine Vergrößerung des rechten Vorhofs in Verbindung mit pulmonaler Hypertonie, wie in Abbildung 6 dargestellt .
7.6 EchokardiographieDie Leitlinie der Europäischen Gesellschaft für Kardiologie (ESC) zur Lungenembolie (PE) empfiehlt eine Echokardiographie als Erstuntersuchung bei Patienten mit anhaltender Dyspnoe, funktionellen Einschränkungen oder Risikofaktoren für CTEPH ( 3 ). Die TTE ist entscheidend für die Abschätzung des pulmonalarteriellen Drucks und bietet wichtige Einblicke in die Pathophysiologie und Ätiologie von CTEPH und die Auswirkungen der chronischen PAH auf das Herz. Sie liefert auch wichtige Daten zu gleichzeitig bestehenden Herzerkrankungen, Operationsergebnissen und möglichen postoperativen Komplikationen wie Tamponade.Bei Patienten mit bestätigter oder stark vermuteter PH ist eine gründliche Untersuchung unerlässlich, um eine linksseitige Herzerkrankung oder intrakardiale Shunts auszuschließen. Marker wie Dilatation und Dysfunktion des rechten Ventrikels gelten als negative Prognoseindikatoren. Darüber hinaus signalisiert ein Perikarderguss im Rahmen einer PH eine fortgeschrittene Erkrankung mit einer schlechten Prognose ( 76 ).Die echokardiographische Bewertung der PH umfasst die Messung der Spitzengeschwindigkeit der Trikuspidalklappeninsuffizienz, die Berechnung des atrioventrikulären Druckgradienten und die Identifizierung indirekter Zeichen der PH wie Dilatation des rechten Vorhofs und Ventrikels, verminderte Kontraktilität des rechten Ventrikels und Doppler-Flussanomalien im Ausflusstrakt des rechten Ventrikels ( 3 ). Diese Indikatoren müssen jedoch in frühen Krankheitsstadien fehlen, und in 10–31 % der Fälle kann die Echokardiographie die PH übersehen ( 77 ).Der erste Schritt zur echokardiografischen Bestimmung der PH-Wahrscheinlichkeit besteht in der Messung der maximalen Trikuspidalinsuffizienzgeschwindigkeit (TRV). Ein Flussdiagramm zur Ermittlung der CTEPH-Wahrscheinlichkeit empfiehlt die Bewertung der Wahrscheinlichkeit für alle klinischen PH-Gruppen unter Einbeziehung zusätzlicher echokardiografischer Parameter, die in drei Kategorien unterteilt sind, die auf PH hinweisen ( 78 ): - Ventrikel: RV/LV-Basaldurchmesser/Flächenverhältnis >1,0; Abflachung des interventrikulären Septums; TAPSE/sPAP-Verhältnis <0,55 mm/mmHg; - Pulmonalarterie: RVOT-Beschleunigungszeit <105 ms und/oder mittsystolisches Notching; frühdiastolische Pulmonalarteriengeschwindigkeit >2,2 m/s; Durchmesser der Pulmonalarterie größer als Durchmesser der Aortenwurzel oder >25 mm; - Untere Hohlvene und rechter Vorhof: Durchmesser der V. cava >21 mm mit verringertem inspiratorischen Kollaps, Fläche des rechten Vorhofs >18 cm2 . Eine TRV > 3,4 m/s deutet auf eine hohe echokardiografische Wahrscheinlichkeit einer PAH hin, während bei einer TRV ≤ 3,4 m/s zusätzliche Parameter aus mindestens zwei Kategorien erforderlich sind, um die Wahrscheinlichkeit einer PAH zu beurteilen ( Abbildung 7 ). Abildung 7
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.
24 Nov 2024 21:34 #2284
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
danny antwortete auf CTEPH, Hintergründe
Zu den weiteren echokardiographischen Markern, die für die Bestimmung der PH-Wahrscheinlichkeit nicht entscheidend, aber für die Beurteilung des Schweregrads und die Bereitstellung von Prognoseinformationen nützlich sind, gehören die Größe des rechten Ventrikels, die fraktionale Flächenänderung, die systolische Exkursion der Trikuspidalringebene (TAPSE), die maximale systolische RV-gepulste Gewebe-Dopplergeschwindigkeit und der rechtsventrikuläre Myokardleistungsindex (RIMP) (
79
).Angesichts der komplexen Geometrie des RV werfen Fortschritte bei Echokardiographietechniken wie Gewebedoppler, 3D-Volumenbildgebung und RV-Belastungsanalyse weiterhin Licht auf die Pathophysiologie der Krankheit.7.7 Lungenfunktionstests und arterielle BlutgaseLungenfunktionstests (LFTs) zur Beurteilung von CTEPH sollten Messungen der gesamten Lungenkapazität und der Diffusionskapazität der Lunge für Kohlenmonoxid (DLCO) umfassen. Bei vielen Patienten mit PAH wird eine leichte restriktive Störung beobachtet, während bei Patienten mit CTEPH häufig eine verringerte DLCO festgestellt wird (
3
). Obwohl LFTs bei CTEPH-Fällen normale Ergebnisse liefern können, können einige Patienten eine leichte Restriktion, eine verringerte Diffusionskapazität oder beides aufweisen.Die Analyse der arteriellen Blutgase (ABG) oder die Entnahme arterialisierter Kapillarblutproben ist entscheidend, um zwischen den verschiedenen PH-Gruppen zu unterscheiden, Begleiterkrankungen zu bewerten und die Notwendigkeit zusätzlicher Sauerstoffzufuhr oder nichtinvasiver Beatmung (NIV) sowie die Schwere der Erkrankung zu bestimmen. Typischerweise weisen Patienten mit PAH normale oder leicht reduzierte PaO2-Werte auf. Ein deutlicher Abfall des PaO2 kann auf ein offenes Foramen ovale, eine Lebererkrankung, andere angeborene Herzfehler mit Rechts-Links-Shunt oder Erkrankungen im Zusammenhang mit niedriger DLCO hinweisen. Der arterielle Kohlendioxidpartialdruck (PaCO2) ist aufgrund der alveolären Hyperventilation bei PAH-Patienten oft niedriger als normal und ist normalerweise mit einer schlechten Prognose verbunden.7.8 Röntgen-ThoraxRöntgenaufnahmen des Brustbereichs sind in den Frühstadien von CTEPH zwar oft nicht aufschlussreich, können aber im Verlauf der Erkrankung Merkmale wie eine Erweiterung der zentralen Lungenarterien und der rechten Herzkammern oder eine Asymmetrie der Lungengefäße nachweisen (
80
). Darüber hinaus kann eine Röntgenaufnahme des Brustbereichs wertvolle Erkenntnisse für die Diagnose anderer Lungenparenchym- oder Herzerkrankungen liefern, einschließlich einer Hypertrophie, die einer CTEPH zugrunde liegen kann. Die wichtigsten auf Röntgenaufnahmen des Brustbereichs erkennbaren Befunde sind in
Tabelle 2
zusammengestellt . Es ist jedoch wichtig anzumerken, dass eine normale Röntgenaufnahme des Brustbereichs das Vorhandensein von PH nicht ausschließt (
81
).
7.9 Ventilations-/Perfusions-LungenszintigraphieDie Ventilations-/Perfusionsszintigraphie (V/Q-Szintigraphie) ist nach wie vor von zentraler Bedeutung bei der Beurteilung von Patienten mit Verdacht auf CTEPH und dient als wichtiges nichtinvasives Diagnoseinstrument zur Erkennung einer pulmonalen Gefäßerkrankung. Das Kennzeichen einer CTEPH bei einem V/Q-Scan ist die Identifizierung anhaltender, nicht übereinstimmender Perfusionsdefekte, die leicht erkennbar sind. Normale V/Q-Scan-Ergebnisse schließen CTEPH effektiv aus und weisen eine hohe Sensitivität von 96–97 % und eine Spezifität von 90–95 % auf ( 82 ).Bei CTED werden typischerweise ein oder mehrere segmentale oder größere Perfusionsdefekte beobachtet. Es ist jedoch wichtig zu beachten, dass die Scan-Befunde das Ausmaß der Lungenarterienobstruktion möglicherweise nicht vollständig erfassen, insbesondere in Fällen mit rekanalisierten Verschlüssen, bei denen die distale Perfusion wiederhergestellt wurde.Die Verwendung der Einzelphotonen-Emissionscomputertomographie (SPECT) V′/Q′-Scanning gegenüber der planaren Bildgebung wird zur Diagnose von APE zunehmend bevorzugt, da sie eine höhere Sensitivität und Spezifität aufweist, insbesondere bei Vorhandensein von Komorbiditäten ( 83 ). Die Anwendung und Validierung dieser Technik speziell für CTEPH ist jedoch noch weniger etabliert ( 81 ).7.10 CT-Untersuchungen des Brustbereichs ohne und mit Kontrastmittel sowie digitale Subtraktionsangiographie der LungeDie CT-Pulmonalisangiographie (CTPA) ist ein wertvolles Diagnoseinstrument zum Ausschluss von CTEPH.Die CTPA kann wertvolle ergänzende Erkenntnisse zu den V/Q-Scans liefern, indem sie eine detaillierte anatomische Lokalisierung ermöglicht und die Durchführbarkeit eines chirurgischen Eingriffs beurteilt ( 84 ). Mit Ausnahme der pulmonalvenookklusiven Krankheit, die sich mit segmentalen Defekten präsentieren kann, führen Erkrankungen des distalen Lungengefäßbetts im Allgemeinen zu normalen Perfusionsscans oder zeigen ein „fleckiges“ Muster, das durch nichtsegmentale Defekte gekennzeichnet ist ( 85 ).In Fällen mit klinischen Hinweisen auf eine akute Lungenembolie kann die CTPA bisher unentdeckte Anzeichen einer CTEPH aufdecken ( 86 ). Sie ist besonders effektiv bei der Identifizierung subsegmentaler oder segmentaler Arterienthrombosen, die für die Bestimmung der chirurgischen Eignung für eine PEA entscheidend sind ( 87 ). Darüber hinaus bietet die hochauflösende Thoraxtomographie (HRCT) wichtige Einblicke in Erkrankungen des Lungenparenchyms wie interstitielle Erkrankungen, Lungenemphysem oder Bronchialanomalien sowie Infarkte und Gefäß- und Thoraxwandfehlbildungen.Trotz ihrer Nützlichkeit ist die CTPA allein kein definitiver Ausschlusskriterium für CTEPH ( 88 ). Sie kann typische Veränderungen, Komorbiditäten und Komplikationen von CTEPH aufdecken, darunter eine Dilatation der Pulmonalarterie, große bronchiale arterielle Kollateralen und die Hypoperfusionsbereiche, die für das „Mosaikmuster“ charakteristisch sind, obwohl dieses Muster auch bei 12 % der Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) auftreten kann ( 89 ). Zu den CT-Befunden, die auf PH hinweisen, zählen ein vergrößerter Durchmesser der Pulmonalarterie (PA), eine Begradigung oder Linksbeugung des Ventrikelseptums, eine Dilatation des rechten Ventrikels (RV) und Hypertrophie.Die CTPA zeigt für CTEPH spezifische Gefäßanomalien, wie z. B. eine Erweiterung der zentralen Lungenarterie mit verringertem Durchmesser der peripheren Gefäße und exzentrische, an der Lungenwand haftende Thromben (möglicherweise verkalkt). Diese Veränderungen unterscheiden sich deutlich von dem für eine akute Lungenembolie typischen zentralen Füllungsdefekt. Eine Dilatation der Bronchialarterien ist zwar nicht nur bei CTEPH typisch, wird aber häufig beobachtet und ist mit besseren Ergebnissen nach PEA verbunden ( 90 ).Die pulmonale digitale Subtraktionsangiographie (PDSA) gilt traditionell als Goldstandard zur Beschreibung der Gefäßmorphologie bei CTEPH. Sie kann Verschlüsse (Pouchdefekte), Netze oder Bänder, unregelmäßige Gefäßwände und eine abrupte Verengung oder ein plötzliches Verschwinden von Gefäßen identifizieren. Während die PDSA nach wie vor von entscheidender Bedeutung ist, insbesondere wenn die CTPA-Ergebnisse nicht eindeutig sind, verändern Fortschritte bei nichtinvasiven Bildgebungsverfahren die diagnostische Landschaft. Selektive segmentale Angiographie, Cone-Beam-CT und Flächendetektor-CT bieten eine verbesserte Visualisierung der subsegmentalen Gefäße und helfen bei der Planung von BPA-Verfahren ( 3 ).Neue Bildgebungsverfahren wie die Magnetresonanz-Pulmonalisangiographie, die Dual-Energy-CT und die Cone-Beam-CT sind vielversprechende Verfahren für eine detaillierte Untersuchung der Lungengefäße ( 91 – 93 ) und bieten umfassende diagnostische Möglichkeiten bei der Beurteilung der CTEPH.7.11 Kardiopulmonale BelastungstestsDer kardiopulmonale Belastungstest (CPET) dient als fortschrittliches Diagnoseinstrument, mit dem durch Untersuchung der Pathophysiologie der Belastungsintoleranz zwischen verschiedenen Ursachen von Dyspnoe differenziert werden kann. Bei Personen mit PH zeigt der CPET deutliche Veränderungen, darunter einen verringerten endtidalen Kohlendioxidpartialdruck (PETCO2), ein erhöhtes Ventilationsäquivalent für Kohlendioxid (VE/VCO2), einen verminderten Sauerstoffpuls (VO2/HR) und eine verringerte maximale Sauerstoffaufnahme (VO2) ( 94 ). Ebenso weisen Personen mit einer Lungengefäßerkrankung ein spezifisches CPET-Muster auf, das durch verringerte körperliche Leistungsfähigkeit, begrenztes Schlagvolumen, erhöhte Totraumventilation und ventilatorische Ineffizienz gekennzeichnet ist ( 95 ). Es wird erwartet, dass der Einsatz von CPET neben der Belastungshämodynamik für die Bewertung und Überwachung von CTEPH ohne PH an Bedeutung gewinnen wird, was einen bedeutenden Fortschritt bei der Diagnose und Nachsorge von CTEPH darstellt.CPET kann bei Patienten mit normaler Ruheechokardiographie subtile Anomalien erkennen, wie z. B. eine erhöhte VE/VCO2-Steigung und ein verringertes PETCO2 an der anaeroben Schwelle, die auf eine Lungengefäßerkrankung hinweisen ( 96 , 97 ). Darüber hinaus haben CPET-Parameter wie maximale Belastungs-VD/VT >45 % eine hohe Sensitivität (92 %) und Spezifität (83 %) zur Vorhersage von CTEPH gezeigt ( 98 ).Die invasive kardiopulmonale Belastungsdiagnostik (iCPET) verbessert die diagnostischen Fähigkeiten der CPET noch weiter, indem sie die zentrale Hämodynamik und den Gasaustausch während der Belastung direkt misst. Bei der iCPET werden Katheter in die rechte Seite des Herzens und der Pulmonalarterie eingeführt, wodurch der pulmonalarterielle Druck (PAP), der PCWP und das Herzzeitvolumen während der Belastung präzise gemessen werden können. Dieser invasive Ansatz kann versteckte Anomalien im PVR und in der Funktion des rechten Ventrikels aufdecken, die mit der nicht-invasiven CPET allein nicht erkennbar sind ( 98 , 99 ). So kann die iCPET zum Beispiel zwischen präkapillären und postkapillären Ursachen der PH unterscheiden und wichtige Erkenntnisse für eine gezielte Behandlung liefern ( 97 , 99 ).Studien haben gezeigt, dass die iCPET besonders bei der Diagnose von CTEPH hilfreich ist, da sie belastungsbedingte PAP- und PVR-Erhöhungen aufdecken kann, die das Vorliegen einer signifikanten Lungengefäßobstruktion bestätigen. Diese detaillierte hämodynamische Beurteilung ist entscheidend, um CTEPH von anderen Formen der PH zu unterscheiden und geeignete therapeutische Interventionen einzuleiten ( 97 , 99 ).Durch die Integration von CPET und iCPET in andere Diagnoseverfahren wie Echokardiographie und Rechtsherzkatheterisierung können Kliniker eine umfassendere Beurteilung der Patienten erreichen, insbesondere derjenigen mit anhaltenden Symptomen nach einer Lungenembolie ( 97 – 99 ).7.12 RechtsherzkatheterEine Rechtsherzkatheterisierung (RHC) ist unverzichtbar, um zu bestätigen, dass die Hämodynamik des Lungenkreislaufs den Diagnosekriterien für PH entspricht: mittlerer Pulmonalarteriendruck (PA) über 20 mmHg und pulmonalarterieller Verschlussdruck (PAWP) von 15 mmHg oder weniger ( 100 ). Die Durchführung einer RHC erfordert Fachwissen und die Einhaltung standardisierter Protokolle zur genauen Messung des Drucks im rechten Vorhof (RA), der rechten Herzkammer (RV) und der Pulmonalarterie sowie zur Bestimmung des Herzzeitvolumens (entweder mit Ficks direkter Methode oder durch Thermodilution, wobei mindestens drei Messungen gemittelt werden) und der gemischtvenösen Blutsättigung. Außerdem wird mit diesem Verfahren der pulmonalvaskuläre Widerstand berechnet, ein entscheidender Prognoseindikator sowohl für prä- als auch postoperative Untersuchungen ( 101 ).Es gibt absolute Kontraindikationen für die Durchführung einer RHC, darunter das Vorhandensein von Thromben oder Tumoren im RA oder RV, vor kurzem implantierte Herzschrittmacher (weniger als einen Monat alt), mechanische Rechtsherzklappen, TriClip-Geräte und akute Infektionen/Endokarditis. Das Nutzen-Risiko-Verhältnis einer RHC muss vor dem Verfahren sorgfältig abgewogen und mit dem Patienten besprochen werden. Obwohl es Komplikationen bei einer RHC gibt, ist ihre Inzidenz erheblich gering, im Allgemeinen weniger als 1 % ( 102 ). Eine der schwerwiegendsten Komplikationen in Verbindung mit einer RHC ist die Perforation der Pulmonalarterie (PA), was die Bedeutung einer gründlichen Patientenvorbereitung unterstreicht. Eine optimale Kontrolle bereits bestehender Erkrankungen, insbesondere von Blutdruck und Volumen, ist zum Zeitpunkt der Untersuchung unverzichtbar.Als Null-Referenzwert für Katheterdruckmessungen, einschließlich PAWP, wird der mittlere Thoraxbereich empfohlen, der bei den meisten Patienten der Position des linken Vorhofs entspricht ( 103 ). Diese Messungen sollten am Ende der Ausatmung vorgenommen werden, um Atemanhaltemanöver zu vermeiden. Bei Patienten mit erheblichen intrathorakalen Druckschwankungen (z. B. bei COPD, Fettleibigkeit oder während körperlicher Belastung) werden jedoch aus Genauigkeitsgründen Durchschnittswerte über drei bis vier Atemzyklen berechnet. Für genaue PAWP-Werte muss der Katheter für Sättigungsmessungen in der Keilposition platziert werden ( 104 ).Die Messung des pulmonalarteriellen Verschlussdrucks (PAOP) ist wichtig, um eine postkapilläre PH aufgrund von Komorbiditäten auszuschließen. In Fällen von CTEPH, bei denen intravaskuläre Obstruktionen die PAOP-Bestimmung erschweren können, sollte der linksventrikuläre enddiastolische Druck mittels Linksventrikelkatheterisierung bestimmt werden ( 96 ). Insbesondere bei zentraler CTEPH kann der Blutdruck aufgrund zentraler organisierender Thromben lokal schwanken, sodass Messungen sowohl in der linken als auch in der rechten Hauptpulmonalarterie und in den PA-Stämmen erforderlich sind. Außerdem kann die Messung des PA-Verschlussdrucks durch die Anhaftung organischer Thromben erschwert werden, sodass vor der Messung eine sorgfältige Wellenformanalyse erforderlich ist ( 105 ). Eine Bewertung der wichtigsten Diagnosemethoden finden Sie in Tabelle 3 .
7.13 DiagnosealgorithmusFür eine genaue und rechtzeitige Diagnose von CTEPH müssen Ärzte bei Patienten mit unerklärlicher Dyspnoe oder Symptomen, die auf eine Rechtsherzinsuffizienz hinweisen, äußerst misstrauisch sein. Eine umfassende Untersuchung unter Einsatz verschiedener multimodaler Bildgebungsverfahren wie Echokardiografie, CT-Scans und Ventilations-/Perfusions-(V/Q-)Scans ist für eine höhere Diagnosegenauigkeit bei CTEPH unerlässlich. In Fällen, in denen CTEPH vermutet wird, sind Rechtsherzkatheterisierung (RHC), CTPA und digitale Subtraktionsangiografie (PDSA) – insbesondere wenn die CTPA-Ergebnisse nicht eindeutig sind – zwingend erforderlich, um die Diagnose zu bestätigen und die therapeutische Entscheidungsfindung zu steuern ( Abbildung 8 ).
8 Screening und FrüherkennungScreening ist ein entscheidender Ansatz zur Identifizierung nicht erkannter Erkrankungen oder Risikomarker und kann sowohl auf Einzelpersonen als auch auf Bevölkerungsgruppen angewendet werden, unabhängig vom Vorhandensein von Symptomen. Diese Methode zielt darauf ab, potenzielle Krankheiten frühzeitig zu erkennen, um rechtzeitig eingreifen und so Morbidität und Mortalität senken zu können. Trotz Fortschritten bei Therapien, die das Fortschreiten von CTEPH und PH verlangsamen, erfolgt die Diagnose häufig mehr als zwei Jahre nach Symptombeginn, was CTEPH als häufig unterdiagnostizierte Lungenembolie-Komplikation hervorhebt. Ein routinemäßiges PH-Screening nach einer Lungenembolie wird nicht allgemein empfohlen, sollte jedoch bei Patienten mit anhaltenden Symptomen in Betracht gezogen werden ( 106 ).Die ESC-Leitlinien von 2019 beschreiben einen Screening-Algorithmus für das Post-PE-Syndrom und empfehlen eine erste Patientenuntersuchung 3–6 Monate nach der PE. Dieser Zeitpunkt steht im Einklang mit der Überprüfung der Antikoagulationstherapie und der Früherkennung von bestehender oder neuer Dyspnoe und/oder funktionellen Einschränkungen ( 57 ). Darüber hinaus sollten Risikofaktoren wie permanente intravaskuläre Geräte und bestimmte Gesundheitszustände berücksichtigt werden ( 24 , 57 ). Ein Screening auf das Antiphospholipid-Syndrom bei der Diagnose von CTEPH wird empfohlen, wobei Entscheidungen zur langfristigen Antikoagulationstherapie von Fall zu Fall getroffen werden ( 73 ).Bei Patienten mit Symptomen oder Risikofaktoren für CTEPH ist die Echokardiographie aufgrund ihrer Sicherheit und Aussagekraft die bevorzugte Diagnosemethode ( 107 ). Die TTE, das erste Screening- und Follow-up-Tool für CTEPH, schätzt den systolischen PA-Druck und untersucht den rechten Ventrikel/Vorhof, Klappenstrukturen, intrakardiale Shunts und andere PH-Ursachen ( 3 , 108 ). Bei geringer PH-Wahrscheinlichkeit sollten andere Dyspnoe-Ursachen untersucht werden. Bei mittlerer Wahrscheinlichkeit können weitere Tests, einschließlich BNP- oder NT-proBNP-Spiegel und CPET, angezeigt sein. Normale NT-proBNP-Spiegel und typische EKG-Befunde der RV-Belastung sagen das Fehlen von CTEPH signifikant voraus ( 109 , 110 ).Die Leiden-Ausschlusskriterien für CTEPH und der InShape-II-Algorithmus sind besonders wichtige Instrumente für die Diagnose von CTEPH. Die Leiden-Ausschlusskriterien für CTEPH verwenden eine Kombination aus EKG- und NT-proBNP-Werten, um CTEPH bei Patienten mit einer Vorgeschichte akuter Lungenembolien und Verdacht auf CTEPH wirksam auszuschließen. Dieses Instrument hat sich als hochsensitiv und spezifitätsstark erwiesen, was es zu einer zuverlässigen Methode macht, um die Krankheit auf nicht-invasive Weise auszuschließen ( 111 ). Der InShape-II-Algorithmus bezieht zudem klinische Parameter, EKG- und NT-proBNP-Werte mit ein und rationalisiert den Diagnoseprozess, indem er klare Kriterien für den Ausschluss von CTEPH liefert und so den Bedarf an invasiveren und kostspieligeren Verfahren minimiert ( 112 ).Bei hoher Erkrankungswahrscheinlichkeit werden zur Erkennung von Perfusionsdefekten nach einer Antikoagulationstherapie ein Lungen-V/Q-Scan oder neuere Methoden wie Jodsubtraktionsmapping, DECT und MRT-Perfusion in Betracht gezogen. Trotz der theoretischen Vorteile dieser neueren Techniken sind sie technisch anspruchsvoll, teurer und es fehlt ihnen an umfassender Validierung ( 3 ). Aufgrund ihrer hohen Sensitivität und Spezifität bleibt die V/Q-Szintigraphie das wichtigste nichtinvasive Screening-Tool für CTEPH ( 3 ). Jüngste Studien untersuchten die Sensitivität und Spezifität neuer CT-Techniken im Vergleich zu herkömmlichen V/Q-Scans und kamen zu dem Ergebnis, dass sie vergleichbar wirksam, jedoch komplexer und weniger zugänglich sind ( 113 , 114 ).9 FazitCTEPH, das sich in etwa 20 % der Fälle aus APE entwickelt, geht in eine chronische Erkrankung über, die durch Gefäßverengung, Gefäßumbau und Proliferation von glatten Muskel- und Endothelzellen gekennzeichnet ist, was zu erhöhtem Druck in der Pulmonalarterie und der rechten Herzkammer und in der Folge zu Rechtsherzversagen führt ( 38 , 96 ). Ziel dieser Übersicht ist es, Klinikern einen umfassenden Leitfaden zur Epidemiologie, Pathophysiologie, zu Risikofaktoren und Diagnostik bei Patienten mit Verdacht auf CTEPH zu bieten.Aufgrund seiner Seltenheit, Letalität und Behinderung muss CTEPH umgehend diagnostiziert und behandelt werden. Für eine frühzeitige Erkennung müssen Ärzte bei Patienten mit unerklärlicher Dyspnoe oder Anzeichen einer rechtsseitigen Herzinsuffizienz einen hohen Verdachtsindex beibehalten und diese gründlich untersuchen. Die Verwendung einer Kombination aus bildgebenden Verfahren wie Echokardiographie, CT und V/Q-Scans ist für eine höhere Diagnosegenauigkeit unerlässlich. Rechtsherzkatheterisierung (RHC) und digitale Subtraktionsangiographie (DSA) sind entscheidend für die Bestätigung der CTEPH-Diagnose und die Entscheidung über die Behandlung.Autorenbeiträge
7.9 Ventilations-/Perfusions-LungenszintigraphieDie Ventilations-/Perfusionsszintigraphie (V/Q-Szintigraphie) ist nach wie vor von zentraler Bedeutung bei der Beurteilung von Patienten mit Verdacht auf CTEPH und dient als wichtiges nichtinvasives Diagnoseinstrument zur Erkennung einer pulmonalen Gefäßerkrankung. Das Kennzeichen einer CTEPH bei einem V/Q-Scan ist die Identifizierung anhaltender, nicht übereinstimmender Perfusionsdefekte, die leicht erkennbar sind. Normale V/Q-Scan-Ergebnisse schließen CTEPH effektiv aus und weisen eine hohe Sensitivität von 96–97 % und eine Spezifität von 90–95 % auf ( 82 ).Bei CTED werden typischerweise ein oder mehrere segmentale oder größere Perfusionsdefekte beobachtet. Es ist jedoch wichtig zu beachten, dass die Scan-Befunde das Ausmaß der Lungenarterienobstruktion möglicherweise nicht vollständig erfassen, insbesondere in Fällen mit rekanalisierten Verschlüssen, bei denen die distale Perfusion wiederhergestellt wurde.Die Verwendung der Einzelphotonen-Emissionscomputertomographie (SPECT) V′/Q′-Scanning gegenüber der planaren Bildgebung wird zur Diagnose von APE zunehmend bevorzugt, da sie eine höhere Sensitivität und Spezifität aufweist, insbesondere bei Vorhandensein von Komorbiditäten ( 83 ). Die Anwendung und Validierung dieser Technik speziell für CTEPH ist jedoch noch weniger etabliert ( 81 ).7.10 CT-Untersuchungen des Brustbereichs ohne und mit Kontrastmittel sowie digitale Subtraktionsangiographie der LungeDie CT-Pulmonalisangiographie (CTPA) ist ein wertvolles Diagnoseinstrument zum Ausschluss von CTEPH.Die CTPA kann wertvolle ergänzende Erkenntnisse zu den V/Q-Scans liefern, indem sie eine detaillierte anatomische Lokalisierung ermöglicht und die Durchführbarkeit eines chirurgischen Eingriffs beurteilt ( 84 ). Mit Ausnahme der pulmonalvenookklusiven Krankheit, die sich mit segmentalen Defekten präsentieren kann, führen Erkrankungen des distalen Lungengefäßbetts im Allgemeinen zu normalen Perfusionsscans oder zeigen ein „fleckiges“ Muster, das durch nichtsegmentale Defekte gekennzeichnet ist ( 85 ).In Fällen mit klinischen Hinweisen auf eine akute Lungenembolie kann die CTPA bisher unentdeckte Anzeichen einer CTEPH aufdecken ( 86 ). Sie ist besonders effektiv bei der Identifizierung subsegmentaler oder segmentaler Arterienthrombosen, die für die Bestimmung der chirurgischen Eignung für eine PEA entscheidend sind ( 87 ). Darüber hinaus bietet die hochauflösende Thoraxtomographie (HRCT) wichtige Einblicke in Erkrankungen des Lungenparenchyms wie interstitielle Erkrankungen, Lungenemphysem oder Bronchialanomalien sowie Infarkte und Gefäß- und Thoraxwandfehlbildungen.Trotz ihrer Nützlichkeit ist die CTPA allein kein definitiver Ausschlusskriterium für CTEPH ( 88 ). Sie kann typische Veränderungen, Komorbiditäten und Komplikationen von CTEPH aufdecken, darunter eine Dilatation der Pulmonalarterie, große bronchiale arterielle Kollateralen und die Hypoperfusionsbereiche, die für das „Mosaikmuster“ charakteristisch sind, obwohl dieses Muster auch bei 12 % der Patienten mit idiopathischer pulmonaler arterieller Hypertonie (IPAH) auftreten kann ( 89 ). Zu den CT-Befunden, die auf PH hinweisen, zählen ein vergrößerter Durchmesser der Pulmonalarterie (PA), eine Begradigung oder Linksbeugung des Ventrikelseptums, eine Dilatation des rechten Ventrikels (RV) und Hypertrophie.Die CTPA zeigt für CTEPH spezifische Gefäßanomalien, wie z. B. eine Erweiterung der zentralen Lungenarterie mit verringertem Durchmesser der peripheren Gefäße und exzentrische, an der Lungenwand haftende Thromben (möglicherweise verkalkt). Diese Veränderungen unterscheiden sich deutlich von dem für eine akute Lungenembolie typischen zentralen Füllungsdefekt. Eine Dilatation der Bronchialarterien ist zwar nicht nur bei CTEPH typisch, wird aber häufig beobachtet und ist mit besseren Ergebnissen nach PEA verbunden ( 90 ).Die pulmonale digitale Subtraktionsangiographie (PDSA) gilt traditionell als Goldstandard zur Beschreibung der Gefäßmorphologie bei CTEPH. Sie kann Verschlüsse (Pouchdefekte), Netze oder Bänder, unregelmäßige Gefäßwände und eine abrupte Verengung oder ein plötzliches Verschwinden von Gefäßen identifizieren. Während die PDSA nach wie vor von entscheidender Bedeutung ist, insbesondere wenn die CTPA-Ergebnisse nicht eindeutig sind, verändern Fortschritte bei nichtinvasiven Bildgebungsverfahren die diagnostische Landschaft. Selektive segmentale Angiographie, Cone-Beam-CT und Flächendetektor-CT bieten eine verbesserte Visualisierung der subsegmentalen Gefäße und helfen bei der Planung von BPA-Verfahren ( 3 ).Neue Bildgebungsverfahren wie die Magnetresonanz-Pulmonalisangiographie, die Dual-Energy-CT und die Cone-Beam-CT sind vielversprechende Verfahren für eine detaillierte Untersuchung der Lungengefäße ( 91 – 93 ) und bieten umfassende diagnostische Möglichkeiten bei der Beurteilung der CTEPH.7.11 Kardiopulmonale BelastungstestsDer kardiopulmonale Belastungstest (CPET) dient als fortschrittliches Diagnoseinstrument, mit dem durch Untersuchung der Pathophysiologie der Belastungsintoleranz zwischen verschiedenen Ursachen von Dyspnoe differenziert werden kann. Bei Personen mit PH zeigt der CPET deutliche Veränderungen, darunter einen verringerten endtidalen Kohlendioxidpartialdruck (PETCO2), ein erhöhtes Ventilationsäquivalent für Kohlendioxid (VE/VCO2), einen verminderten Sauerstoffpuls (VO2/HR) und eine verringerte maximale Sauerstoffaufnahme (VO2) ( 94 ). Ebenso weisen Personen mit einer Lungengefäßerkrankung ein spezifisches CPET-Muster auf, das durch verringerte körperliche Leistungsfähigkeit, begrenztes Schlagvolumen, erhöhte Totraumventilation und ventilatorische Ineffizienz gekennzeichnet ist ( 95 ). Es wird erwartet, dass der Einsatz von CPET neben der Belastungshämodynamik für die Bewertung und Überwachung von CTEPH ohne PH an Bedeutung gewinnen wird, was einen bedeutenden Fortschritt bei der Diagnose und Nachsorge von CTEPH darstellt.CPET kann bei Patienten mit normaler Ruheechokardiographie subtile Anomalien erkennen, wie z. B. eine erhöhte VE/VCO2-Steigung und ein verringertes PETCO2 an der anaeroben Schwelle, die auf eine Lungengefäßerkrankung hinweisen ( 96 , 97 ). Darüber hinaus haben CPET-Parameter wie maximale Belastungs-VD/VT >45 % eine hohe Sensitivität (92 %) und Spezifität (83 %) zur Vorhersage von CTEPH gezeigt ( 98 ).Die invasive kardiopulmonale Belastungsdiagnostik (iCPET) verbessert die diagnostischen Fähigkeiten der CPET noch weiter, indem sie die zentrale Hämodynamik und den Gasaustausch während der Belastung direkt misst. Bei der iCPET werden Katheter in die rechte Seite des Herzens und der Pulmonalarterie eingeführt, wodurch der pulmonalarterielle Druck (PAP), der PCWP und das Herzzeitvolumen während der Belastung präzise gemessen werden können. Dieser invasive Ansatz kann versteckte Anomalien im PVR und in der Funktion des rechten Ventrikels aufdecken, die mit der nicht-invasiven CPET allein nicht erkennbar sind ( 98 , 99 ). So kann die iCPET zum Beispiel zwischen präkapillären und postkapillären Ursachen der PH unterscheiden und wichtige Erkenntnisse für eine gezielte Behandlung liefern ( 97 , 99 ).Studien haben gezeigt, dass die iCPET besonders bei der Diagnose von CTEPH hilfreich ist, da sie belastungsbedingte PAP- und PVR-Erhöhungen aufdecken kann, die das Vorliegen einer signifikanten Lungengefäßobstruktion bestätigen. Diese detaillierte hämodynamische Beurteilung ist entscheidend, um CTEPH von anderen Formen der PH zu unterscheiden und geeignete therapeutische Interventionen einzuleiten ( 97 , 99 ).Durch die Integration von CPET und iCPET in andere Diagnoseverfahren wie Echokardiographie und Rechtsherzkatheterisierung können Kliniker eine umfassendere Beurteilung der Patienten erreichen, insbesondere derjenigen mit anhaltenden Symptomen nach einer Lungenembolie ( 97 – 99 ).7.12 RechtsherzkatheterEine Rechtsherzkatheterisierung (RHC) ist unverzichtbar, um zu bestätigen, dass die Hämodynamik des Lungenkreislaufs den Diagnosekriterien für PH entspricht: mittlerer Pulmonalarteriendruck (PA) über 20 mmHg und pulmonalarterieller Verschlussdruck (PAWP) von 15 mmHg oder weniger ( 100 ). Die Durchführung einer RHC erfordert Fachwissen und die Einhaltung standardisierter Protokolle zur genauen Messung des Drucks im rechten Vorhof (RA), der rechten Herzkammer (RV) und der Pulmonalarterie sowie zur Bestimmung des Herzzeitvolumens (entweder mit Ficks direkter Methode oder durch Thermodilution, wobei mindestens drei Messungen gemittelt werden) und der gemischtvenösen Blutsättigung. Außerdem wird mit diesem Verfahren der pulmonalvaskuläre Widerstand berechnet, ein entscheidender Prognoseindikator sowohl für prä- als auch postoperative Untersuchungen ( 101 ).Es gibt absolute Kontraindikationen für die Durchführung einer RHC, darunter das Vorhandensein von Thromben oder Tumoren im RA oder RV, vor kurzem implantierte Herzschrittmacher (weniger als einen Monat alt), mechanische Rechtsherzklappen, TriClip-Geräte und akute Infektionen/Endokarditis. Das Nutzen-Risiko-Verhältnis einer RHC muss vor dem Verfahren sorgfältig abgewogen und mit dem Patienten besprochen werden. Obwohl es Komplikationen bei einer RHC gibt, ist ihre Inzidenz erheblich gering, im Allgemeinen weniger als 1 % ( 102 ). Eine der schwerwiegendsten Komplikationen in Verbindung mit einer RHC ist die Perforation der Pulmonalarterie (PA), was die Bedeutung einer gründlichen Patientenvorbereitung unterstreicht. Eine optimale Kontrolle bereits bestehender Erkrankungen, insbesondere von Blutdruck und Volumen, ist zum Zeitpunkt der Untersuchung unverzichtbar.Als Null-Referenzwert für Katheterdruckmessungen, einschließlich PAWP, wird der mittlere Thoraxbereich empfohlen, der bei den meisten Patienten der Position des linken Vorhofs entspricht ( 103 ). Diese Messungen sollten am Ende der Ausatmung vorgenommen werden, um Atemanhaltemanöver zu vermeiden. Bei Patienten mit erheblichen intrathorakalen Druckschwankungen (z. B. bei COPD, Fettleibigkeit oder während körperlicher Belastung) werden jedoch aus Genauigkeitsgründen Durchschnittswerte über drei bis vier Atemzyklen berechnet. Für genaue PAWP-Werte muss der Katheter für Sättigungsmessungen in der Keilposition platziert werden ( 104 ).Die Messung des pulmonalarteriellen Verschlussdrucks (PAOP) ist wichtig, um eine postkapilläre PH aufgrund von Komorbiditäten auszuschließen. In Fällen von CTEPH, bei denen intravaskuläre Obstruktionen die PAOP-Bestimmung erschweren können, sollte der linksventrikuläre enddiastolische Druck mittels Linksventrikelkatheterisierung bestimmt werden ( 96 ). Insbesondere bei zentraler CTEPH kann der Blutdruck aufgrund zentraler organisierender Thromben lokal schwanken, sodass Messungen sowohl in der linken als auch in der rechten Hauptpulmonalarterie und in den PA-Stämmen erforderlich sind. Außerdem kann die Messung des PA-Verschlussdrucks durch die Anhaftung organischer Thromben erschwert werden, sodass vor der Messung eine sorgfältige Wellenformanalyse erforderlich ist ( 105 ). Eine Bewertung der wichtigsten Diagnosemethoden finden Sie in Tabelle 3 .
7.13 DiagnosealgorithmusFür eine genaue und rechtzeitige Diagnose von CTEPH müssen Ärzte bei Patienten mit unerklärlicher Dyspnoe oder Symptomen, die auf eine Rechtsherzinsuffizienz hinweisen, äußerst misstrauisch sein. Eine umfassende Untersuchung unter Einsatz verschiedener multimodaler Bildgebungsverfahren wie Echokardiografie, CT-Scans und Ventilations-/Perfusions-(V/Q-)Scans ist für eine höhere Diagnosegenauigkeit bei CTEPH unerlässlich. In Fällen, in denen CTEPH vermutet wird, sind Rechtsherzkatheterisierung (RHC), CTPA und digitale Subtraktionsangiografie (PDSA) – insbesondere wenn die CTPA-Ergebnisse nicht eindeutig sind – zwingend erforderlich, um die Diagnose zu bestätigen und die therapeutische Entscheidungsfindung zu steuern ( Abbildung 8 ).
8 Screening und FrüherkennungScreening ist ein entscheidender Ansatz zur Identifizierung nicht erkannter Erkrankungen oder Risikomarker und kann sowohl auf Einzelpersonen als auch auf Bevölkerungsgruppen angewendet werden, unabhängig vom Vorhandensein von Symptomen. Diese Methode zielt darauf ab, potenzielle Krankheiten frühzeitig zu erkennen, um rechtzeitig eingreifen und so Morbidität und Mortalität senken zu können. Trotz Fortschritten bei Therapien, die das Fortschreiten von CTEPH und PH verlangsamen, erfolgt die Diagnose häufig mehr als zwei Jahre nach Symptombeginn, was CTEPH als häufig unterdiagnostizierte Lungenembolie-Komplikation hervorhebt. Ein routinemäßiges PH-Screening nach einer Lungenembolie wird nicht allgemein empfohlen, sollte jedoch bei Patienten mit anhaltenden Symptomen in Betracht gezogen werden ( 106 ).Die ESC-Leitlinien von 2019 beschreiben einen Screening-Algorithmus für das Post-PE-Syndrom und empfehlen eine erste Patientenuntersuchung 3–6 Monate nach der PE. Dieser Zeitpunkt steht im Einklang mit der Überprüfung der Antikoagulationstherapie und der Früherkennung von bestehender oder neuer Dyspnoe und/oder funktionellen Einschränkungen ( 57 ). Darüber hinaus sollten Risikofaktoren wie permanente intravaskuläre Geräte und bestimmte Gesundheitszustände berücksichtigt werden ( 24 , 57 ). Ein Screening auf das Antiphospholipid-Syndrom bei der Diagnose von CTEPH wird empfohlen, wobei Entscheidungen zur langfristigen Antikoagulationstherapie von Fall zu Fall getroffen werden ( 73 ).Bei Patienten mit Symptomen oder Risikofaktoren für CTEPH ist die Echokardiographie aufgrund ihrer Sicherheit und Aussagekraft die bevorzugte Diagnosemethode ( 107 ). Die TTE, das erste Screening- und Follow-up-Tool für CTEPH, schätzt den systolischen PA-Druck und untersucht den rechten Ventrikel/Vorhof, Klappenstrukturen, intrakardiale Shunts und andere PH-Ursachen ( 3 , 108 ). Bei geringer PH-Wahrscheinlichkeit sollten andere Dyspnoe-Ursachen untersucht werden. Bei mittlerer Wahrscheinlichkeit können weitere Tests, einschließlich BNP- oder NT-proBNP-Spiegel und CPET, angezeigt sein. Normale NT-proBNP-Spiegel und typische EKG-Befunde der RV-Belastung sagen das Fehlen von CTEPH signifikant voraus ( 109 , 110 ).Die Leiden-Ausschlusskriterien für CTEPH und der InShape-II-Algorithmus sind besonders wichtige Instrumente für die Diagnose von CTEPH. Die Leiden-Ausschlusskriterien für CTEPH verwenden eine Kombination aus EKG- und NT-proBNP-Werten, um CTEPH bei Patienten mit einer Vorgeschichte akuter Lungenembolien und Verdacht auf CTEPH wirksam auszuschließen. Dieses Instrument hat sich als hochsensitiv und spezifitätsstark erwiesen, was es zu einer zuverlässigen Methode macht, um die Krankheit auf nicht-invasive Weise auszuschließen ( 111 ). Der InShape-II-Algorithmus bezieht zudem klinische Parameter, EKG- und NT-proBNP-Werte mit ein und rationalisiert den Diagnoseprozess, indem er klare Kriterien für den Ausschluss von CTEPH liefert und so den Bedarf an invasiveren und kostspieligeren Verfahren minimiert ( 112 ).Bei hoher Erkrankungswahrscheinlichkeit werden zur Erkennung von Perfusionsdefekten nach einer Antikoagulationstherapie ein Lungen-V/Q-Scan oder neuere Methoden wie Jodsubtraktionsmapping, DECT und MRT-Perfusion in Betracht gezogen. Trotz der theoretischen Vorteile dieser neueren Techniken sind sie technisch anspruchsvoll, teurer und es fehlt ihnen an umfassender Validierung ( 3 ). Aufgrund ihrer hohen Sensitivität und Spezifität bleibt die V/Q-Szintigraphie das wichtigste nichtinvasive Screening-Tool für CTEPH ( 3 ). Jüngste Studien untersuchten die Sensitivität und Spezifität neuer CT-Techniken im Vergleich zu herkömmlichen V/Q-Scans und kamen zu dem Ergebnis, dass sie vergleichbar wirksam, jedoch komplexer und weniger zugänglich sind ( 113 , 114 ).9 FazitCTEPH, das sich in etwa 20 % der Fälle aus APE entwickelt, geht in eine chronische Erkrankung über, die durch Gefäßverengung, Gefäßumbau und Proliferation von glatten Muskel- und Endothelzellen gekennzeichnet ist, was zu erhöhtem Druck in der Pulmonalarterie und der rechten Herzkammer und in der Folge zu Rechtsherzversagen führt ( 38 , 96 ). Ziel dieser Übersicht ist es, Klinikern einen umfassenden Leitfaden zur Epidemiologie, Pathophysiologie, zu Risikofaktoren und Diagnostik bei Patienten mit Verdacht auf CTEPH zu bieten.Aufgrund seiner Seltenheit, Letalität und Behinderung muss CTEPH umgehend diagnostiziert und behandelt werden. Für eine frühzeitige Erkennung müssen Ärzte bei Patienten mit unerklärlicher Dyspnoe oder Anzeichen einer rechtsseitigen Herzinsuffizienz einen hohen Verdachtsindex beibehalten und diese gründlich untersuchen. Die Verwendung einer Kombination aus bildgebenden Verfahren wie Echokardiographie, CT und V/Q-Scans ist für eine höhere Diagnosegenauigkeit unerlässlich. Rechtsherzkatheterisierung (RHC) und digitale Subtraktionsangiographie (DSA) sind entscheidend für die Bestätigung der CTEPH-Diagnose und die Entscheidung über die Behandlung.Autorenbeiträge
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.