
- Beiträge: 1757
Sidebar
PAH bei angeborenen Herzfehler
16 Nov 2024 22:30 #2269
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
PAH bei angeborenen Herzfehler wurde erstellt von danny
www.frontiersin.org/journals/cardiovascu...360555/fullPatienten mit pulmonaler Hypertonie in Verbindung mit einem Links-Rechts-Shunt weisen ein breites Spektrum pathophysiologischer Substrate auf, von Patienten mit pulmonaler Überzirkulation bis hin zu Patienten mit fortgeschrittener pulmonaler Gefäßerkrankung. Die erste Gruppe kann in sorgfältig ausgewählten Fällen von einer Shunt-Reparatur profitieren, aber wenn sich bereits eine fortgeschrittene pulmonale Gefäßerkrankung entwickelt hat, sollte ein Defektverschluss vermieden werden und pulmonale Vasodilatatoren können eingesetzt werden, um die Belastungstoleranz und Hämodynamik zu verbessern. Es gibt jedoch nur wenige Belege, die die Entscheidungsfindung bei der Behandlung dieser Patienten unterstützen. Wir diskutieren die Behandlungsgrundsätze bei Patienten mit pulmonaler Hypertonie und einem vorherrschenden Links-Rechts-Shunt. Die in diesem Artikel gemachten Empfehlungen und Aussagen basieren auf pathophysiologischen Überlegungen und Expertenmeinungen. 1 EinleitungDaten aus klinischen Studien haben die langfristige Wirksamkeit von Therapien gegen pulmonale arterielle Hypertonie (PAH) bei bestimmten Patientenkohorten mit PAH in Verbindung mit angeborenem Herzfehler (AHF) klar belegt (
1
). Es muss jedoch betont werden, dass es sich bei PAH in Verbindung mit AHF (PAH-AHF) um eine sehr heterogene Population handelt, mit großen Unterschieden in Bezug auf die kardiovaskuläre Pathophysiologie und Prognose zwischen den 4 Hauptuntergruppen: Eisenmenger-Syndrom, PAH und Links-Rechts-Shunt, PAH mit kleinem/zufälligem Shunt und PAH nach Shunt-Reparatur eines AHF (
2
,
3
).Heutzutage gibt es genügend Belege, die den Einsatz von PAH-Therapien bei Patienten mit Eisenmenger-Syndrom unterstützen, da sie zu einer Verbesserung der Belastungstoleranz, der Funktionsklasse und der pulmonalen Hämodynamik führen (
4
–
7
). PAH-Therapien werden auch routinemäßig bei Patienten mit Anzeichen einer pulmonalen Gefäßerkrankung (PVD) nach Shunt-Reparatur eingesetzt. Tatsächlich lässt sich bei bis zu 5 – 10 % der operierten Patienten eine pulmonale Hypertonie feststellen (
8
), die hinsichtlich Prognose und Therapieansprechen oft einen klinischen Phänotyp aufweisen, der dem der idiopathischen PAH ähnelt (
3
,
9
). In beiden Kohorten verringert ein Abfall des pulmonal-vaskulären Widerstands (PVR) und eine Steigerung des pulmonalen Blutflusses im Ruhezustand und insbesondere bei Anstrengung die Nachlast des rechten Ventrikels (RV) und optimiert die Sauerstoffzufuhr zum peripheren Gewebe.Der potenzielle Nutzen von PAH-Therapien für Patienten mit leichter oder mittelschwerer PAH in Verbindung mit einem Links-Rechts-Shunt ist weniger klar, und es fehlen Belege. Bei Patienten mit leichteren Formen von PAH kann eine Reparatur durchgeführt werden, während es für die übrigen Patienten nur wenige Belege für die Behandlung gibt. Trotz der spärlichen Datenlage erhält ein erheblicher Anteil dieser Patienten PAH-Therapien in spezialisierten Zentren (
10
). Ziel dieser Übersichtsarbeit ist es, die einzigartige und dynamische Pathophysiologie der PAH in Verbindung mit einem Links-Rechts-Shunt zu bewerten und praktische Behandlungsmethoden für diese wichtige Untergruppe von Patienten mit angeborener Herzkrankheit zu diskutieren.2 EpidemiologieObwohl die Inzidenz der PAH-Analoga in den Industrieländern zurückgeht, ist sie weltweit immer noch weit verbreitet und betrifft zwischen 4 % und 28 % der Erwachsenen mit angeborenem Herzfehler (
9
,
10
). Eine genaue Schätzung der Prävalenz der PAH-Analoga mit einem Links-Rechts-Shunt ist besonders schwierig, da sich nur wenige Berichte auf diese Bevölkerungsgruppe konzentrieren (
11
).Im CONCOR-CHD-Register hatten 15 von 135 PAH-CHD-Patienten einen Links-Rechts-Shunt; nur eine Minderheit dieser Patienten erhielt PAH-Therapien (
8
). In einem anderen großen, multizentrischen PAH-CHD-Register waren ein Viertel der Patienten Nicht-Eisenmenger-Patienten (167/680, 24 %) und alle erhielten eine zielgerichtete Therapie (
9
). Diese Zahlen schließen jedoch Patienten aus den drei Nicht-Eisenmenger-Kategorien ein (Links-Rechts-Shunt, zufälliger Shunt und nach Reparatur) und sollten mit Vorsicht interpretiert werden. Tatsächlich stellen Patienten mit PAH und Links-Rechts-Shunts eine eigenständige Entität dar, für deren Prävalenz und Behandlung es nur wenige Belege gibt, weshalb sie in der klinischen Praxis empirisch behandelt werden. Es ist auch wichtig zu bedenken, dass sich bei vielen dieser Patienten im Laufe der Zeit ein Eisenmenger-Syndrom entwickeln kann oder sie in die Gruppe nach Reparatur wechseln, wenn sie sich einer perkutanen oder chirurgischen Reparatur unterziehen und sich herausstellt, dass sie eine Rest-PAH haben.3 PathophysiologieSeit Paul Woods Abhandlung über die Pathophysiologie des Eisenmenger-Syndroms in den 1950er Jahren (
12
) wurden viele Hypothesen zur Entwicklung der PVD bei Patienten mit angeborenen Shunts aufgestellt. Inzwischen gilt als anerkannt, dass es sich bei der PVD um ein Kontinuum anatomischer und pathophysiologischer Veränderungen handelt, die je nach Art und Schwere des Shunts letztendlich zu einer irreversiblen Erkrankung führen können. Verschiedene genetische, physiologische und anatomische Variablen können die Entwicklung einer fortgeschrittenen PVD modulieren, wobei das Zeitfenster für die Reparatur des Defekts von Person zu Person sehr unterschiedlich sein kann (
13
). Es gibt auch kleine Kohorten von Kleinkindern, bei denen sich der Lungenkreislauf nach der Geburt nicht angemessen umgestaltet und histologische Veränderungen in der Lunge, wie z. B. die Intimaproliferation, zu früh im Leben auftreten, als dass sie wesentlich durch die kumulative Scherspannung der Lungengefäße beeinflusst werden könnten (
14
).Bei Patienten, die aufgrund einer nachgewiesenen PVD als irreparabel gelten, wird häufig eine PAH-Therapie in Betracht gezogen, obwohl die Rationalität des Einsatzes pulmonaler Vasodilatatoren bei einem beträchtlichen Links-Rechts-Shunt seit einiger Zeit diskutiert wird (
15
). Zudem unterscheiden sich die klinischen und hämodynamischen Ziele der PAH-Therapie bei dieser Patientengruppe häufig von denen bei anderen Untergruppen der PAH-CHD.3.1 Genetische ModulatorenDie Rolle genetischer Faktoren bei der Modulation des Beginns und der Schwere der Erkrankung bei idiopathischer PAH ist mittlerweile bekannt. Mutationen im Gen des Bone Morphogenetic Protein Receptor II ( BMPR2 ) sind am häufigsten (
16
). Kürzlich wurde eine Rolle pathogenetischer Polymorphismen bei PAH-CHD vorgeschlagen: BMPR2- Mutationen wurden bei 6 % der Erwachsenen und Kinder mit PAH-CHD (hauptsächlich beim Eisenmenger-Syndrom oder nach der Reparatur) festgestellt, obwohl die Rolle von BMPR2 bei PAH-CHD unklar bleibt (
17
). Eine erhöhte Prävalenz von Mutationen in den Genen des SRY-Box-Transkriptionsfaktors 17 ( SOX-17 ) und des T-Box-Transkriptionsfaktor-Gens ( TBX4 ) wurde bei Patienten festgestellt, die nach der Reparatur eines Ventrikelseptumdefekts eine PAH entwickelten, sowie bei Patienten mit prätrikuspiden Shunts und Eisenmenger-Physiologie (
18
-
20
). Andere Gene ( ABCC8 und SMAD1 ) wurden bei Patienten mit PAH in Verbindung mit einem kleinen/zufälligen Defekt beschrieben (
18
). Genetische Faktoren könnten die Variabilität bei der Entwicklung einer PVD bei Patienten mit prätrikuspidalen Shunts erklären, bei denen der erhöhte Lungenblutfluss allein als unzureichend für die Entwicklung einer PVD angesehen wird (
21
).Die molekularen Mechanismen, die den Beginn und das Fortschreiten der PAH beeinflussen, wurden weiter untersucht. Dies legt nahe, dass epigenetische Faktoren eine wichtige Rolle bei der Gestaltung der klinischen Ausprägung der verschiedenen genetischen Varianten spielen. Diese Faktoren, zu denen Sequenzen nicht-kodierender RNA, Mikro-RNA und Proteinpolymorphismen gehören, sind noch immer schlecht verstanden, und eine umfassende Bewertung dieses Themas geht über den Rahmen dieser Übersicht hinaus (
22
). Jüngste mechanistische Arbeiten haben eine Verbindung zwischen Signalen, die die Zellproliferation und -differenzierung regulieren, und der Biologie der glatten Lungenmuskelzellen gezeigt (
23
). Verschiedene Expressionsgrade von Proteinen, die an der Proliferation glatter Muskelzellen beteiligt sind, korrelieren mit dem Schweregrad der PVD in Tiermodellen der flussassoziierten PAH; solche Proteine können eine schützende Rolle gegen vaskuläre Umgestaltung bei Vorhandensein eines Links-Rechts-Shunts spielen (
23
). Diese Daten werfen neues Licht auf die Pathogenese der PAH-CHD und entfernen sich von der traditionellen Ansicht, dass dies ein Zustand ist, der ausschließlich durch die Hämodynamik ausgelöst wird. Genetische Faktoren spielen wahrscheinlich eine Rolle dabei, ob sich eine PVD entwickelt (insbesondere bei Patienten mit prätrikuspidalen Shunts) und wie schnell sie fortschreitet, was sich auf die Operabilität, das Ansprechen auf PAH-Therapien und die Prognose in dieser Kohorte auswirkt.3.2 Hämodynamische ModulatorenShunts auf verschiedenen anatomischen Ebenen bergen unterschiedliche Risiken für PVD. Große posttrikuspidale Shunts führen zu Druck- und Volumenbelastung des Lungenkreislaufs und verursachen viel häufiger PVD als prätrikuspidale Shunts (z. B. Vorhofseptumdefekte). Dies gilt insbesondere für angeborene Shunts zwischen den großen Arterien wie dem gemeinsamen Arterienstamm, dem aortopulmonalen Fenster oder einem großen offenen Ductus arteriosus, die während des gesamten Herzzyklus eine erhebliche Scherspannung im Lungenkreislauf erzeugen. Bleiben sie unbehandelt, verursachen sie wahrscheinlich eine schwere PVD und entwickeln sich schon früh im Leben zu einer Eisenmenger-Physiologie (
12
). Wagenvoort et al. wurde gezeigt, dass die Bänderung der Pulmonalarterien bei Patienten mit posttrikuspidalen Shunts und histologischen Hinweisen auf eine PVD die Krankheit bei einigen Patienten rückgängig machte, höchstwahrscheinlich bei Patienten mit leichteren Formen der PVD. Dies verdeutlichte das Spektrum der Schwere der Erkrankung und die Schwierigkeiten bei der Feststellung, bei wem eine irreversible pulmonale Arteriopathie aufgetreten ist, mittels Herzkatheterisierung oder sogar Lungenbiopsie (
24
,
25
).Bei der Beurteilung des pulmonalen Blutflusses und des Shuntanteils müssen verschiedene Faktoren berücksichtigt werden. Bei prätrikuspidalen Shunts hängt der Shuntanteil nicht nur von der Größe des Defekts ab, sondern auch vom Druckgradienten zwischen den Vorhöfen. Erkrankungen, die den Druck im linken Vorhof erhöhen, wie z. B. eine Erkrankung der linksseitigen Herzklappe oder eine systemische ventrikuläre Funktionsstörung, fördern wahrscheinlich einen Links-Rechts-Shunt, während Erkrankungen, die den Druck im rechten Vorhof erhöhen, wie z. B. eine Erkrankung der Trikuspidalklappe, eine Obstruktion des rechtsventrikulären Ausflusstrakts oder pulmonale Hypertonie, den Links-Rechts-Shunt verringern und eine Shunt-Umkehr verursachen können (
Abbildung 1
). Abildung 1
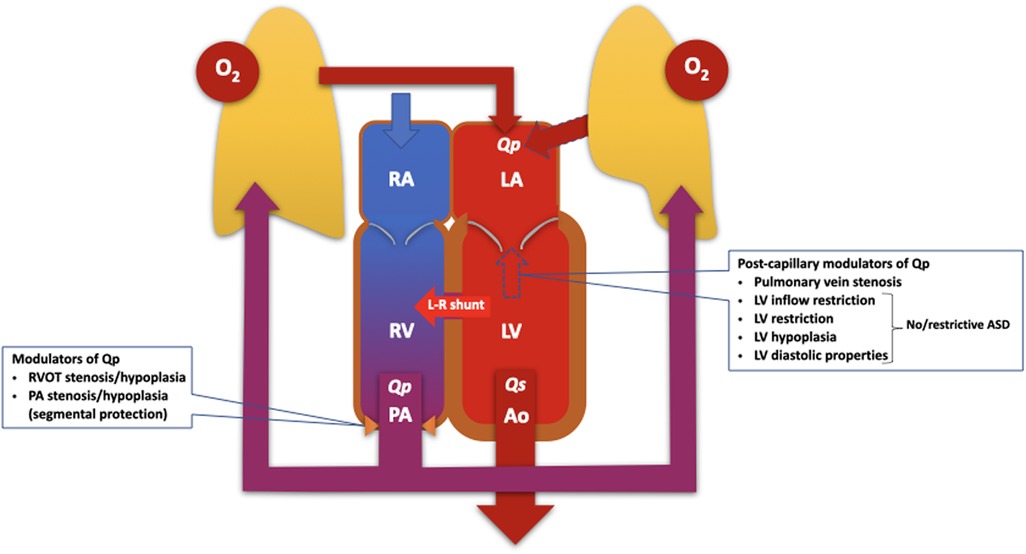 Abbildung 1 . Diagramm zur Veranschaulichung der Pathophysiologie des Lungen- und Systemkreislaufs, die parallel arbeiten, um den Lungenblutfluss und die Schwere des Links-Rechts-Shunts (LR) zu modulieren. Die Modulatoren des Lungenblutflusses und der Shunt-Traktion können grundsätzlich als prä- und postkapillär eingeteilt werden und sollten bei der Beurteilung von Patienten mit PAH und Links-Rechts-Shunts berücksichtigt werden. ASD, Vorhofseptumdefekt; LA, linker Vorhof; LV, linker Ventrikel/ventrikulär; O2 , Sauerstoff; PA, Lungenarterie; Qp, Lungenblutfluss; Qs, systemischer Blutfluss; RA, rechter Vorhof; RV, rechter Ventrikel; RVOT, rechtsventrikulärer Ausflusstrakt. Die Pathophysiologie wird noch komplizierter, wenn eine segmentale pulmonale Hypertonie vorliegt, das heißt, wenn sich eine PVD in einigen, aber nicht in allen Lungensegmenten entwickelt (
26
). Dies ist typisch für Patienten mit Fallot-Tetralogie und Pulmonalatresie, kann aber auch bei Patienten mit einem großen posttrikuspidalen Shunt und einer Stenose der Pulmonalarterienäste auftreten.Gelegentlich zeigen Patienten mit großen, nicht restriktiven posttrikuspidalen Verbindungen erst spät eine überraschend leichte PAH und einen vorherrschenden Links-Rechts-Shunt, mit oder ohne Anzeichen einer Lungenstauung. In Abwesenheit einer Obstruktion des rechtsventrikulären Ausflusstrakts oder einer Stenose eines Lungenarterienastes, die das Lungengefäßbett schützen könnte, wird bei solchen Patienten ein „nachgiebiger“ Lungenkreislauf angenommen, der weniger empfindlich auf Scherspannungen und die durch den Shunt verursachte Überlastung reagiert. Ob dieser Phänotyp genetisch bedingt ist, muss noch ermittelt werden.
Abbildung 1 . Diagramm zur Veranschaulichung der Pathophysiologie des Lungen- und Systemkreislaufs, die parallel arbeiten, um den Lungenblutfluss und die Schwere des Links-Rechts-Shunts (LR) zu modulieren. Die Modulatoren des Lungenblutflusses und der Shunt-Traktion können grundsätzlich als prä- und postkapillär eingeteilt werden und sollten bei der Beurteilung von Patienten mit PAH und Links-Rechts-Shunts berücksichtigt werden. ASD, Vorhofseptumdefekt; LA, linker Vorhof; LV, linker Ventrikel/ventrikulär; O2 , Sauerstoff; PA, Lungenarterie; Qp, Lungenblutfluss; Qs, systemischer Blutfluss; RA, rechter Vorhof; RV, rechter Ventrikel; RVOT, rechtsventrikulärer Ausflusstrakt. Die Pathophysiologie wird noch komplizierter, wenn eine segmentale pulmonale Hypertonie vorliegt, das heißt, wenn sich eine PVD in einigen, aber nicht in allen Lungensegmenten entwickelt (
26
). Dies ist typisch für Patienten mit Fallot-Tetralogie und Pulmonalatresie, kann aber auch bei Patienten mit einem großen posttrikuspidalen Shunt und einer Stenose der Pulmonalarterienäste auftreten.Gelegentlich zeigen Patienten mit großen, nicht restriktiven posttrikuspidalen Verbindungen erst spät eine überraschend leichte PAH und einen vorherrschenden Links-Rechts-Shunt, mit oder ohne Anzeichen einer Lungenstauung. In Abwesenheit einer Obstruktion des rechtsventrikulären Ausflusstrakts oder einer Stenose eines Lungenarterienastes, die das Lungengefäßbett schützen könnte, wird bei solchen Patienten ein „nachgiebiger“ Lungenkreislauf angenommen, der weniger empfindlich auf Scherspannungen und die durch den Shunt verursachte Überlastung reagiert. Ob dieser Phänotyp genetisch bedingt ist, muss noch ermittelt werden.
4 Behandlung von PAH bei Links-Rechts-ShuntsEs gibt sehr wenige Belege für den Einsatz von PAH-Therapien bei Patienten mit angeborener Herzkrankheit und vorherrschendem Links-Rechts-Shunt ( 1 , 27 – 31 ). In anderen PAH-Kohorten werden diese Therapien eingesetzt, um den PVR zu senken und den pulmonalen Blutfluss in Ruhe und bei Anstrengung zu steigern, wodurch die RV-Nachlast reduziert und die Kopplung zwischen RV und Lungenkreislauf verbessert wird. Bei Patienten mit leichter PAH und einem beträchtlichen Links-Rechts-Shunt (d. h. Verhältnis von pulmonalem zu systemischem Blutfluss oder Qp/Qs > 1,5) steigern PAH-Therapien wahrscheinlich den bereits übermäßigen pulmonalen Blutfluss weiter und verschlimmern so potenziell die Überlastung des rechten oder linken Ventrikels und des Lungenkreislaufs. In solchen Fällen ist eine PAH-Therapie nur sinnvoll, um den PVR auf ein Niveau zu senken, das eine Reparatur des Defekts ermöglicht (der sogenannte „Treat-and-Repair“-Ansatz, der erstmals von Dimopoulos et al. vorgeschlagen wurde) ( 32 ). Der Einsatz der Dreifachkombinationstherapie, ein Eckpfeiler in der Behandlung der schweren idiopathischen PAH, ist bei dieser Patientengruppe nicht nachweisbar. Patienten mit vorherrschenden Links-Rechts-Shunts werden in PAH-CHD-Kohorten, in denen die Auswirkungen einer Kombinationstherapie untersucht werden, häufig ausgeschlossen oder sind unterrepräsentiert ( 33 , 34 ). Vor und nach Beginn der PAH-Therapie ist eine Herzkatheterisierung erforderlich, um den Rückgang des PVR auf Werte zu dokumentieren, die einen teilweisen (oder vollständigen) Verschluss des Defekts ermöglichen und so die ventrikuläre Überlastung beseitigen und das Risiko einer Progression der PVD verringern. Die aktuellen ESC-Leitlinien zur angeborenen Herzkrankheit bei Erwachsenen enthalten klare Empfehlungen für eine Behandlungs- und Reparaturstrategie für Patienten mit einem Vorhofseptumdefekt und einem Ausgangs-PVR ≥ 5 WU ( 35 ). Sie empfehlen einen fenestrierten Verschluss des Defekts nach der PAH-Therapie, wenn der PVR auf < 5 WU abfällt und Hinweise auf eine RV-Volumenüberlastung mit einem Qp/Qs > 1,5 vorliegen. Es ist erwähnenswert, dass dieser Grenzwert relativ konservativ ist; in der Vergangenheit wurden Vorhofseptumdefekte mit einem PVR von 6–8 WU geschlossen ( 36 ). Der fenestrierte Verschluss von prätrikuspidalen Shunts bei Patienten mit PAH mit gleichzeitiger Einleitung einer PAH-Therapie zum Zeitpunkt der Reparatur wurde beschrieben, obwohl dies nicht in gleicher Weise wie die „Treat-and-Repair“-Methode in den ACHD-Leitlinien der ESC ( 35 , 37 ) befürwortet wird. Tatsächlich wird ein signifikanter Abfall des PVR nach der Therapie als wesentlich erachtet, um einen Defektverschluss in Betracht zu ziehen, und es besteht das Risiko, dass Patienten, die langfristig nicht ausreichend auf PAH-Therapien ansprechen, selbst durch einen teilweisen Verschluss des Defekts negativ beeinflusst werden können. Es wird im Allgemeinen erwartet, dass PAH-Therapien bei Patienten, bei denen eine erfolgreiche Behandlung und Reparatur durchgeführt wurde (die jetzt zur PAH-CHD-Untergruppe nach der Reparatur gehören), auf unbestimmte Zeit verschrieben werden sollten. Nach der Reparatur werden jedoch eine Herzkatheterisierung und eine engmaschige Überwachung mit entsprechender Anpassung der Therapien empfohlen.Bezüglich posttrikuspidaler Links-Rechts-Shunts mit einem PVR ≥ 5 WU gibt es sehr wenige Hinweise zur Behandlung, und Leitlinien empfehlen eine Überweisung an Expertenzentren, die einen ähnlichen Behandlungs- und Reparaturansatz in Erwägung ziehen können. Expertenzentren können sich dafür entscheiden, bei symptomatischen Patienten eine PAH-Therapie (als Einzel- oder Kombinationstherapie) zu beginnen und zu steigern und dabei die Hämodynamik auf einen signifikanten Abfall des PVR und/oder eine signifikante Zunahme des Links-Rechts-Shunts und der ventrikulären Überlastung zu überwachen. Es gibt mehrere Fallberichte und Fallserien von Patienten mit posttrikuspidalen Shunts und etablierter PVD, die auf eine PAH-Therapie ansprachen und bei denen der Defekt repariert wurde ( 38 , 39 ). Patienten mit posttrikuspidalen Shunts weisen jedoch häufig eine schwerere PVD auf und haben einen höheren PVR-Basiswert als ihre Patienten mit prätrikuspidalen Shunts, wodurch eine Behandlungs- und Reparaturstrategie weniger attraktiv wird. Darüber hinaus beziehen sich viele Berichte über erfolgreiche Behandlung und Reparatur von posttrikuspidalen Shunts auf Patienten aus Asien, und ein günstigerer genetischer Hintergrund kann nicht ausgeschlossen werden. Schließlich stellen die Veränderungen der Kriterien für die hämodynamische Operabilität im Laufe der Zeit, Unterschiede im Konsens zwischen analogen Leitlinien und die Variabilität der Behandlung je nach Zentrum und Region eine Herausforderung für die Verwendung von klinischen Daten aus der „realen Welt“ zur Ableitung der Sicherheit dar ( 35 , 40 ). Das Ausmaß des PVR-Abfalls, der mit der PAH-Therapie bei posttrikuspidalen Shunts erreicht wird, variiert erheblich, zwischen 20 % und 40 %, und reproduziert damit in etwa die relative Verringerung, die im interventionellen Arm der klinischen Studien zu idiopathischer PAH berichtet wurde ( 31 , 41 – 43 ). Die Dauer der Behandlung vor der Reparatur mit PAH-Therapien ist ebenfalls nicht standardisiert ( 36 , 38 , 44 ). Es ist wahrscheinlich, dass die meisten Patienten nach der Reparatur eine Langzeitbehandlung benötigen, obwohl dies von der Hämodynamik nach der Reparatur abhängt.Die erforderliche Aufmerksamkeit und Expertise bei der Durchführung und Interpretation einer Herzkatheteruntersuchung bei solchen Patienten kann nicht genug betont werden. Qualitätskontrolle ist unerlässlich, und Fallstricke müssen vermieden werden, z. B. beim Vorhandensein mehrerer Quellen pulmonalen Blutflusses (offener Ductus arteriosus, aortopulmonale Kollateralen), bei den Positionen der Blutentnahme zur Berechnung der gemischten venösen Sättigung und bei der Wahl der verwendeten Gleichung, bei der Verwendung von Sedierung oder Anästhesie, die die Hämodynamik beeinflussen kann, bei der Verabreichung hoher Sauerstoffdosen, die die Shunt-Berechnungen verfälschen können, usw. Eine akute Vasodilatator-Provokation wird derzeit in den Leitlinien nicht befürwortet, kann aber Informationen zur Compliance des Lungengefäßsystems liefern ( 1 , 35 ).Während die kurzfristigen Ergebnisse nach Behandlung und Reparatur in Fallberichten und Fallserien vielversprechend waren, bleiben die langfristigen Auswirkungen des Defektverschlusses bei diesen Patienten ungewiss ( 4 , 30 ). Eine zusätzliche Komplikation bei der Behandlung von PAH-CHD-Patienten mit einem Links-Rechts-Shunt ist der Mangel an Daten zur Risikostratifizierung, die die Grundlage für die Entscheidungsfindung in anderen PAH-Kohorten bildet. Frühere Versionen der ESC/ERS-Leitlinien zur pulmonalen Hypertonie schlugen ein erweitertes dreischichtiges Prognosemodell für PAH vor, das die Behandlung leiten sollte, aber für CHD, insbesondere bei Patienten mit Links-Rechts-Shunts, nicht validiert ist ( 29 ). Dieses Modell wurde im Laufe der Jahre verfeinert und es wurden aktualisierte Prognosemodelle auf der Grundlage europäischer und US-amerikanischer Kohorten entwickelt. In den neuesten Leitlinien von 2022 wurde ein vereinfachtes vierschichtiges Modell vorgeschlagen, das 3 klinische Parameter verwendet (WHO-Funktionsklasse, 6-Minuten-Gehstrecke und bran-natriuretisches Peptid); Die beiden wichtigsten Studien, die dieses Modell stützen, schlossen nur sehr wenige Patienten mit angeborener Herzkrankheit ein, die nicht dem Eisenmenger-Modell unterlagen ( 1 , 28 ). Dieses Modell wurde bei Patienten mit angeborener Herzkrankheit (PAH) angewendet, aber nicht formal validiert ( 45 ).5 Vorschlag für ein praktisches VorgehenBei der Erwägung von PAH-Therapien bei Patienten mit PAH-Angeborenen Herzfehlern und Links-Rechts-Shunts können Expertenzentren die folgenden zwei zentralen Fragen stellen:1. Was sind die Therapieziele und welche klinischen Endpunkte werden zur Beurteilung der Wirksamkeit festgelegt?2. Wie sollte man Patienten auswählen, die am wahrscheinlichsten von einer PAH-Therapie profitieren?Die möglichen Ziele der PAH-Therapie in dieser Kohorte sind entweder:- Erreichen der Operabilitätskriterien, d. h. Modifizierung der pulmonalen Hämodynamik, um die empfohlenen Kriterien für eine Reparatur zu erfüllen (als Teil eines Treat-and-Repair-Ansatzes) ( 35 ), oder- eine Verbesserung des Funktionsstatus, der körperlichen Leistungsfähigkeit und möglicherweise der Prognose bewirken.Defekte, die perkutan repariert werden können, sind für einen Behandlungs- und Reparaturansatz attraktiver, da Operationen, die einen kardiopulmonalen Bypass erfordern, erhebliche Risiken bergen (z. B. perioperative pulmonale Hypertoniekrise), selbst bei Patienten unter wirksamer PAH-Therapie. Eine engmaschige perioperative Überwachung ist unerlässlich, um perioperative Komplikationen zu minimieren, mit einer niedrigen Hemmschwelle für eine Eskalation der PAH-Therapien und hämodynamischen Unterstützung, falls erforderlich. Bei einem Patienten, der nun als PAH-CHD nach der Reparatur klassifiziert wird, sind wiederholte invasive hämodynamische Untersuchungen erforderlich, um die Hämodynamik nach der Reparatur festzustellen und über die Intensität der PAH-Therapie zu entscheiden, mit dem Ziel, die Langzeitergebnisse zu optimieren. Unter diesen Umständen ist die Entscheidung, eine oder mehrere Therapien bei einem Patienten mit normalem oder nahezu normalem PVR abzubrechen, schwierig, und große Vorsicht ist geboten, da die PVD höchstwahrscheinlich bestehen bleibt und sich verschlimmern kann.Bei Patienten mit schwererer PVD, aber immer noch mit vorherrschendem Links-Rechts-Shunt (d. h., die die Eisenmenger-Kriterien noch nicht erfüllen), ist eine Behandlungs- und Reparaturstrategie weniger wahrscheinlich erfolgreich; die Reparatur eines Defekts bei einem Patienten, der eine sehr aggressive PAH-Therapie benötigt, um die hämodynamischen Kriterien für die Operabilität zu erreichen, ist möglicherweise nicht ratsam, und der Nutzen der Beseitigung des Links-Rechts-Shunts überwiegt möglicherweise nicht die Risiken des Eingriffs und die Wahrscheinlichkeit einer fortschreitenden schweren PVD nach der Reparatur des Defekts. Es kann jedoch sinnvoll sein, pulmonale Vasodilatatoren zu verwenden, um die ventrikulovaskuläre und Gasaustauschkopplung zu verbessern, das RV-Schlagvolumen zu optimieren und den pulmonalen Blutfluss durch eine bessere Verteilung auf relativ normale V/Q-Einheiten zu verbessern ( 46 ). In diesem Zusammenhang können PAH-Therapien den PVR und die RV-Nachlast senken, obwohl ein Anstieg des Links-Rechts-Shunts und der ventrikulären Volumenüberlastung wahrscheinlich ist. Darüber hinaus ist unklar, wie diese Strategie die natürliche Entwicklung hin zur Eisenmenger-Physiologie beeinflussen (beschleunigen oder verzögern) könnte.Daher ist es sinnvoll, Patienten mit PAH-Angeborenen Herzfehlern und einem Links-Rechts-Shunt in zwei unterschiedliche Gruppen zu unterteilen, vorausgesetzt, es liegt ein großer prä- oder posttrikuspidaler Defekt vor und es liegen keine Erkrankungen des linken Herzens oder damit verbundene Läsionen vor ( Abbildung 2 ):- Der überwiegend reversible PVD-Typ bezieht sich auf Patienten mit einem großen Links-Rechts-Shunt und Anzeichen von typischerweise leichter pulmonaler Hypertonie. Die überwiegende Mehrheit dieser Patienten gilt als operabel, einige fallen jedoch in eine „Grauzone“ der Entscheidungsfindung, die einer sorgfältigen Expertenbeurteilung bedarf. In dieser Gruppe können PAH-Therapien als Teil eines Behandlungs- und Reparaturansatzes eingesetzt werden, obwohl dies nur ausdrücklich für Vorhofseptumdefekte empfohlen wird. Sobald die Reparatur erreicht ist, werden PAH-Therapien typischerweise langfristig fortgesetzt, obwohl je nach Hämodynamik unmittelbar nach dem Eingriff und langfristig eine Anpassung erforderlich sein kann. Patienten, bei denen Behandlung und Reparatur aufgrund eines unzureichenden Abfalls des PVR trotz angemessener Therapien fehlschlagen, haben wahrscheinlich eine überwiegend irreversible Komponente und sollten als solche klassifiziert werden (siehe unten).- Bei der überwiegend irreversiblen PVD oder dem Prä-Eisenmenger -Typ ist der Links-Rechts-Shunt kleiner als für die Größe des Defekts erwartet, aber es liegt weder in Ruhe noch bei Belastung eine Zyanose vor. In dieser Gruppe können PAH-Therapien unter strenger Überwachung in Betracht gezogen werden, um die Hämodynamik und die RV-Funktion zu verbessern, obwohl es keine Beweise dafür gibt. Der mögliche Nutzen von PAH-Therapien muss gegen das Risiko einer Vergrößerung des Links-Rechts-Shunts und eines RV-Versagens aufgrund einer sich verschlechternden Volumenüberlastung abgewogen werden. Bei diesen Patienten ist es unwahrscheinlich, dass die PAH-Therapie zu einem ausreichenden Abfall des PVR führt, der eine Schließung des Defekts ermöglichen würde. Abildung 2
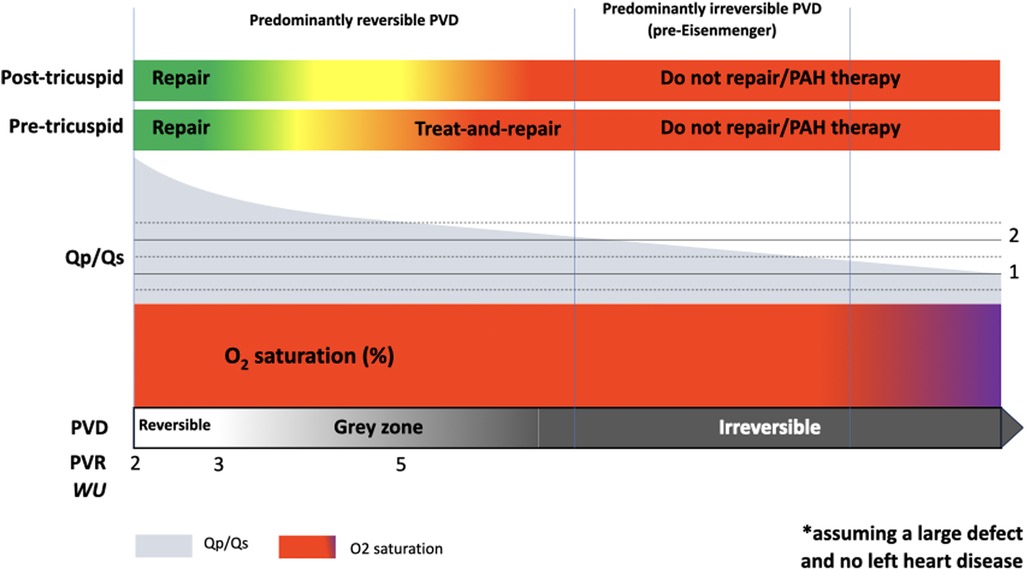 Abbildung 2. Grafische Zusammenfassung der PAH-CHD mit einem Links-Rechts-Shunt, vom überwiegend reversiblen Ende des Spektrums mit leicht erhöhtem pulmonal-vaskulären Widerstand (PVR) und hohem Shunt-Anteil (Qp/Qs) bis zum überwiegend irreversiblen Ende der pulmonal-vaskulären Erkrankung (PVD) oder dem Ende vor Eisenmenger. Dieses Schema geht von einem großen angeborenen Herzfehler und keiner signifikanten Linksherzerkrankung aus. Obwohl Patienten mit PAH-CHD und einem vorherrschenden Links-Rechts-Shunt häufig sind, erfordert eine systematische Beschreibung des Krankheitsspektrums dieser Gruppe eine Rückkehr zu den anatomischen, pathophysiologischen, histologischen und hämodynamischen Konzepten der reversiblen vs. irreversiblen PVD. Leider ist uns das Konzept der Reversibilität trotz jahrzehntelanger Arbeit – 70 Jahre seit der systematischen Beschreibung der Eisenmenger-Physiologie durch Wood und fast 40 Jahre seit der Pionierarbeit von Wagenvoort et al. – immer noch nicht klar (
12
,
25
). Dies beeinflusst unsere Entscheidungsfindung sowohl hinsichtlich der Operabilität als auch des Einsatzes von PAH-Therapien. Dennoch gibt es ein Spektrum innerhalb von PAH-CHD mit einem Links-Rechts-Shunt, das nur im Hinblick auf den Schweregrad der PVD und seine Reversibilität vollständig konzeptualisiert werden kann, d. h. ob unsere Interventionen sich wahrscheinlich günstig auf den natürlichen Verlauf dieser fortschreitenden, lebensbegrenzenden Erkrankung auswirken. Leider lässt sich der Übergang von einer überwiegend reversiblen zu einer überwiegend irreversiblen Erkrankung auf Grundlage der aktuellen Studienlage nicht einfach definieren, was sich in den jüngsten klinischen Empfehlungen widerspiegelt: Die ESC-Leitlinien verwenden <5 WU für die Definition der Operabilität und somit der Reversibilität, erkennen jedoch die erheblichen Unsicherheiten und Variabilität in der Praxis an und empfehlen eine sorgfältige individuelle Beurteilung von Patienten mit einem posttrikuspidalen Shunt und einem PVR ≥ 5 WU in Expertenzentren (
35
). Die Einteilung von Kindern in überwiegend reversible und irreversible PVD ist zwar klinisch wichtig, wird jedoch durch die dynamische Natur der Lungengefäße in der frühen Kindheit und ihre variable Reaktion auf große Shunts (verfälscht durch genetische und andere Faktoren) erheblich erschwert. Auf den physiologischen postnatalen Abfall des PVR folgt typischerweise die Entstehung eines Überzirkulationsphänomens. Dieser ist durch eine Herzinsuffizienz gekennzeichnet, die durch den großen Links-Rechts-Shunt bedingt ist und auf ein noch nachgiebiges Lungengefäßbett hinweist. Es gibt jedoch auch Säuglinge, bei denen der erwartete Abfall des PVR nach der Geburt ausbleibt und die somit nie ein „Fenster der Operabilität“ erleben (
14
).Da für diese Patientenkohorte keine Modelle zur Risikostratifizierung verfügbar sind, ist es sinnvoll, standardmäßige klinische Parameter zu überwachen, wie z. B. WHO-Funktionsklasse, 6-Minuten-Gehstrecke, natriuretische Peptidwerte und echokardiographische Indizes für RV-Dysfunktion und Ankopplung an den Lungenkreislauf. Die kardiale Magnetresonanztomographie kann äußerst hilfreich sein, um die RV-Funktion zu überwachen und den Lungenblutfluss und die Shunt-Fraktion nicht-invasiv zu messen. Der kardiopulmonale Belastungstest (CPET) ist für Patienten, die diesen Test durchführen können (d. h. ohne stark eingeschränkte körperliche Leistungsfähigkeit oder erhebliche Lernschwäche), eine sichere und nützliche nicht-invasive Methode zur Beurteilung der funktionellen Leistungsfähigkeit. Er ist Teil der in den Leitlinien empfohlenen anfänglichen Risikobewertung von Patienten mit PAH (
1
). Die Entwicklung einer progressiven Sauerstoffentsättigung bei Anstrengung kann darauf hindeuten, dass sich ein Patient der Eisenmenger-Physiologie nähert, während Verbesserungen der Sauerstoffsättigung und der Belastungsparameter nach einer PAH-Therapie ein Ansprechen auf die Behandlung anzeigen. Ventilatorische Ineffizienz und Ventilations-Perfusions-Missverhältnis, bestimmt durch ein erhöhtes Verhältnis zwischen Ventilation und CO2 - Produktion (VE/VCO2 - Steigung), stehen in Zusammenhang mit dem Ausgang bei PAH- und nicht-zyanotischen KHK-Kohorten (
47
–
49
). Dies kann zusammen mit anderen Markern der funktionellen Kapazität zur Risikostratifizierung verwendet werden. Eine Rechtsherzkatheterisierung vor und nach Beginn der Therapien ist entscheidend für die Überwachung der Hämodynamik (z. B. Shunt-Fraktion und PVR) und zur Identifizierung von Patienten, die die Operativitätskriterien erreicht haben oder von einer Eskalation der Therapie profitieren könnten. Bei Patienten, die für eine Reparatur nicht in Frage kommen, sollte ein Gleichgewicht zwischen der Optimierung der pulmonalen Hämodynamik, der RV-Nachlast und des Sauerstoffaustauschs angestrebt werden, während gleichzeitig eine signifikante Erhöhung von Qp/Qs vermieden werden sollte, die sich nachteilig auf den RV oder den linken Ventrikel auswirken könnte (bei prä- bzw. posttrikuspidalen Shunts) (
Abbildung 2
).SchlussfolgerungenDie Behandlung von Patienten mit PAH und einem Links-Rechts-Shunt ist besonders anspruchsvoll, da diese Gruppe Personen mit einem breiten Spektrum an hämodynamischen Zuständen umfasst und die Evidenz äußerst begrenzt ist. Mögliche Indikationen für PAH-Therapien unterscheiden sich je nach zugrunde liegender Anatomie und Hämodynamik, wobei zwei wichtige klinische Phänotypen auftreten: überwiegend irreversibler ( vor Eisenmenger ) vs. überwiegend reversibler Typ. Ein maßgeschneiderter fachkundiger Ansatz ist obligatorisch, mit enger Überwachung nach Defektbehebung und/oder Einsatz von PAH-Therapien.
Abbildung 2. Grafische Zusammenfassung der PAH-CHD mit einem Links-Rechts-Shunt, vom überwiegend reversiblen Ende des Spektrums mit leicht erhöhtem pulmonal-vaskulären Widerstand (PVR) und hohem Shunt-Anteil (Qp/Qs) bis zum überwiegend irreversiblen Ende der pulmonal-vaskulären Erkrankung (PVD) oder dem Ende vor Eisenmenger. Dieses Schema geht von einem großen angeborenen Herzfehler und keiner signifikanten Linksherzerkrankung aus. Obwohl Patienten mit PAH-CHD und einem vorherrschenden Links-Rechts-Shunt häufig sind, erfordert eine systematische Beschreibung des Krankheitsspektrums dieser Gruppe eine Rückkehr zu den anatomischen, pathophysiologischen, histologischen und hämodynamischen Konzepten der reversiblen vs. irreversiblen PVD. Leider ist uns das Konzept der Reversibilität trotz jahrzehntelanger Arbeit – 70 Jahre seit der systematischen Beschreibung der Eisenmenger-Physiologie durch Wood und fast 40 Jahre seit der Pionierarbeit von Wagenvoort et al. – immer noch nicht klar (
12
,
25
). Dies beeinflusst unsere Entscheidungsfindung sowohl hinsichtlich der Operabilität als auch des Einsatzes von PAH-Therapien. Dennoch gibt es ein Spektrum innerhalb von PAH-CHD mit einem Links-Rechts-Shunt, das nur im Hinblick auf den Schweregrad der PVD und seine Reversibilität vollständig konzeptualisiert werden kann, d. h. ob unsere Interventionen sich wahrscheinlich günstig auf den natürlichen Verlauf dieser fortschreitenden, lebensbegrenzenden Erkrankung auswirken. Leider lässt sich der Übergang von einer überwiegend reversiblen zu einer überwiegend irreversiblen Erkrankung auf Grundlage der aktuellen Studienlage nicht einfach definieren, was sich in den jüngsten klinischen Empfehlungen widerspiegelt: Die ESC-Leitlinien verwenden <5 WU für die Definition der Operabilität und somit der Reversibilität, erkennen jedoch die erheblichen Unsicherheiten und Variabilität in der Praxis an und empfehlen eine sorgfältige individuelle Beurteilung von Patienten mit einem posttrikuspidalen Shunt und einem PVR ≥ 5 WU in Expertenzentren (
35
). Die Einteilung von Kindern in überwiegend reversible und irreversible PVD ist zwar klinisch wichtig, wird jedoch durch die dynamische Natur der Lungengefäße in der frühen Kindheit und ihre variable Reaktion auf große Shunts (verfälscht durch genetische und andere Faktoren) erheblich erschwert. Auf den physiologischen postnatalen Abfall des PVR folgt typischerweise die Entstehung eines Überzirkulationsphänomens. Dieser ist durch eine Herzinsuffizienz gekennzeichnet, die durch den großen Links-Rechts-Shunt bedingt ist und auf ein noch nachgiebiges Lungengefäßbett hinweist. Es gibt jedoch auch Säuglinge, bei denen der erwartete Abfall des PVR nach der Geburt ausbleibt und die somit nie ein „Fenster der Operabilität“ erleben (
14
).Da für diese Patientenkohorte keine Modelle zur Risikostratifizierung verfügbar sind, ist es sinnvoll, standardmäßige klinische Parameter zu überwachen, wie z. B. WHO-Funktionsklasse, 6-Minuten-Gehstrecke, natriuretische Peptidwerte und echokardiographische Indizes für RV-Dysfunktion und Ankopplung an den Lungenkreislauf. Die kardiale Magnetresonanztomographie kann äußerst hilfreich sein, um die RV-Funktion zu überwachen und den Lungenblutfluss und die Shunt-Fraktion nicht-invasiv zu messen. Der kardiopulmonale Belastungstest (CPET) ist für Patienten, die diesen Test durchführen können (d. h. ohne stark eingeschränkte körperliche Leistungsfähigkeit oder erhebliche Lernschwäche), eine sichere und nützliche nicht-invasive Methode zur Beurteilung der funktionellen Leistungsfähigkeit. Er ist Teil der in den Leitlinien empfohlenen anfänglichen Risikobewertung von Patienten mit PAH (
1
). Die Entwicklung einer progressiven Sauerstoffentsättigung bei Anstrengung kann darauf hindeuten, dass sich ein Patient der Eisenmenger-Physiologie nähert, während Verbesserungen der Sauerstoffsättigung und der Belastungsparameter nach einer PAH-Therapie ein Ansprechen auf die Behandlung anzeigen. Ventilatorische Ineffizienz und Ventilations-Perfusions-Missverhältnis, bestimmt durch ein erhöhtes Verhältnis zwischen Ventilation und CO2 - Produktion (VE/VCO2 - Steigung), stehen in Zusammenhang mit dem Ausgang bei PAH- und nicht-zyanotischen KHK-Kohorten (
47
–
49
). Dies kann zusammen mit anderen Markern der funktionellen Kapazität zur Risikostratifizierung verwendet werden. Eine Rechtsherzkatheterisierung vor und nach Beginn der Therapien ist entscheidend für die Überwachung der Hämodynamik (z. B. Shunt-Fraktion und PVR) und zur Identifizierung von Patienten, die die Operativitätskriterien erreicht haben oder von einer Eskalation der Therapie profitieren könnten. Bei Patienten, die für eine Reparatur nicht in Frage kommen, sollte ein Gleichgewicht zwischen der Optimierung der pulmonalen Hämodynamik, der RV-Nachlast und des Sauerstoffaustauschs angestrebt werden, während gleichzeitig eine signifikante Erhöhung von Qp/Qs vermieden werden sollte, die sich nachteilig auf den RV oder den linken Ventrikel auswirken könnte (bei prä- bzw. posttrikuspidalen Shunts) (
Abbildung 2
).SchlussfolgerungenDie Behandlung von Patienten mit PAH und einem Links-Rechts-Shunt ist besonders anspruchsvoll, da diese Gruppe Personen mit einem breiten Spektrum an hämodynamischen Zuständen umfasst und die Evidenz äußerst begrenzt ist. Mögliche Indikationen für PAH-Therapien unterscheiden sich je nach zugrunde liegender Anatomie und Hämodynamik, wobei zwei wichtige klinische Phänotypen auftreten: überwiegend irreversibler ( vor Eisenmenger ) vs. überwiegend reversibler Typ. Ein maßgeschneiderter fachkundiger Ansatz ist obligatorisch, mit enger Überwachung nach Defektbehebung und/oder Einsatz von PAH-Therapien.
4 Behandlung von PAH bei Links-Rechts-ShuntsEs gibt sehr wenige Belege für den Einsatz von PAH-Therapien bei Patienten mit angeborener Herzkrankheit und vorherrschendem Links-Rechts-Shunt ( 1 , 27 – 31 ). In anderen PAH-Kohorten werden diese Therapien eingesetzt, um den PVR zu senken und den pulmonalen Blutfluss in Ruhe und bei Anstrengung zu steigern, wodurch die RV-Nachlast reduziert und die Kopplung zwischen RV und Lungenkreislauf verbessert wird. Bei Patienten mit leichter PAH und einem beträchtlichen Links-Rechts-Shunt (d. h. Verhältnis von pulmonalem zu systemischem Blutfluss oder Qp/Qs > 1,5) steigern PAH-Therapien wahrscheinlich den bereits übermäßigen pulmonalen Blutfluss weiter und verschlimmern so potenziell die Überlastung des rechten oder linken Ventrikels und des Lungenkreislaufs. In solchen Fällen ist eine PAH-Therapie nur sinnvoll, um den PVR auf ein Niveau zu senken, das eine Reparatur des Defekts ermöglicht (der sogenannte „Treat-and-Repair“-Ansatz, der erstmals von Dimopoulos et al. vorgeschlagen wurde) ( 32 ). Der Einsatz der Dreifachkombinationstherapie, ein Eckpfeiler in der Behandlung der schweren idiopathischen PAH, ist bei dieser Patientengruppe nicht nachweisbar. Patienten mit vorherrschenden Links-Rechts-Shunts werden in PAH-CHD-Kohorten, in denen die Auswirkungen einer Kombinationstherapie untersucht werden, häufig ausgeschlossen oder sind unterrepräsentiert ( 33 , 34 ). Vor und nach Beginn der PAH-Therapie ist eine Herzkatheterisierung erforderlich, um den Rückgang des PVR auf Werte zu dokumentieren, die einen teilweisen (oder vollständigen) Verschluss des Defekts ermöglichen und so die ventrikuläre Überlastung beseitigen und das Risiko einer Progression der PVD verringern. Die aktuellen ESC-Leitlinien zur angeborenen Herzkrankheit bei Erwachsenen enthalten klare Empfehlungen für eine Behandlungs- und Reparaturstrategie für Patienten mit einem Vorhofseptumdefekt und einem Ausgangs-PVR ≥ 5 WU ( 35 ). Sie empfehlen einen fenestrierten Verschluss des Defekts nach der PAH-Therapie, wenn der PVR auf < 5 WU abfällt und Hinweise auf eine RV-Volumenüberlastung mit einem Qp/Qs > 1,5 vorliegen. Es ist erwähnenswert, dass dieser Grenzwert relativ konservativ ist; in der Vergangenheit wurden Vorhofseptumdefekte mit einem PVR von 6–8 WU geschlossen ( 36 ). Der fenestrierte Verschluss von prätrikuspidalen Shunts bei Patienten mit PAH mit gleichzeitiger Einleitung einer PAH-Therapie zum Zeitpunkt der Reparatur wurde beschrieben, obwohl dies nicht in gleicher Weise wie die „Treat-and-Repair“-Methode in den ACHD-Leitlinien der ESC ( 35 , 37 ) befürwortet wird. Tatsächlich wird ein signifikanter Abfall des PVR nach der Therapie als wesentlich erachtet, um einen Defektverschluss in Betracht zu ziehen, und es besteht das Risiko, dass Patienten, die langfristig nicht ausreichend auf PAH-Therapien ansprechen, selbst durch einen teilweisen Verschluss des Defekts negativ beeinflusst werden können. Es wird im Allgemeinen erwartet, dass PAH-Therapien bei Patienten, bei denen eine erfolgreiche Behandlung und Reparatur durchgeführt wurde (die jetzt zur PAH-CHD-Untergruppe nach der Reparatur gehören), auf unbestimmte Zeit verschrieben werden sollten. Nach der Reparatur werden jedoch eine Herzkatheterisierung und eine engmaschige Überwachung mit entsprechender Anpassung der Therapien empfohlen.Bezüglich posttrikuspidaler Links-Rechts-Shunts mit einem PVR ≥ 5 WU gibt es sehr wenige Hinweise zur Behandlung, und Leitlinien empfehlen eine Überweisung an Expertenzentren, die einen ähnlichen Behandlungs- und Reparaturansatz in Erwägung ziehen können. Expertenzentren können sich dafür entscheiden, bei symptomatischen Patienten eine PAH-Therapie (als Einzel- oder Kombinationstherapie) zu beginnen und zu steigern und dabei die Hämodynamik auf einen signifikanten Abfall des PVR und/oder eine signifikante Zunahme des Links-Rechts-Shunts und der ventrikulären Überlastung zu überwachen. Es gibt mehrere Fallberichte und Fallserien von Patienten mit posttrikuspidalen Shunts und etablierter PVD, die auf eine PAH-Therapie ansprachen und bei denen der Defekt repariert wurde ( 38 , 39 ). Patienten mit posttrikuspidalen Shunts weisen jedoch häufig eine schwerere PVD auf und haben einen höheren PVR-Basiswert als ihre Patienten mit prätrikuspidalen Shunts, wodurch eine Behandlungs- und Reparaturstrategie weniger attraktiv wird. Darüber hinaus beziehen sich viele Berichte über erfolgreiche Behandlung und Reparatur von posttrikuspidalen Shunts auf Patienten aus Asien, und ein günstigerer genetischer Hintergrund kann nicht ausgeschlossen werden. Schließlich stellen die Veränderungen der Kriterien für die hämodynamische Operabilität im Laufe der Zeit, Unterschiede im Konsens zwischen analogen Leitlinien und die Variabilität der Behandlung je nach Zentrum und Region eine Herausforderung für die Verwendung von klinischen Daten aus der „realen Welt“ zur Ableitung der Sicherheit dar ( 35 , 40 ). Das Ausmaß des PVR-Abfalls, der mit der PAH-Therapie bei posttrikuspidalen Shunts erreicht wird, variiert erheblich, zwischen 20 % und 40 %, und reproduziert damit in etwa die relative Verringerung, die im interventionellen Arm der klinischen Studien zu idiopathischer PAH berichtet wurde ( 31 , 41 – 43 ). Die Dauer der Behandlung vor der Reparatur mit PAH-Therapien ist ebenfalls nicht standardisiert ( 36 , 38 , 44 ). Es ist wahrscheinlich, dass die meisten Patienten nach der Reparatur eine Langzeitbehandlung benötigen, obwohl dies von der Hämodynamik nach der Reparatur abhängt.Die erforderliche Aufmerksamkeit und Expertise bei der Durchführung und Interpretation einer Herzkatheteruntersuchung bei solchen Patienten kann nicht genug betont werden. Qualitätskontrolle ist unerlässlich, und Fallstricke müssen vermieden werden, z. B. beim Vorhandensein mehrerer Quellen pulmonalen Blutflusses (offener Ductus arteriosus, aortopulmonale Kollateralen), bei den Positionen der Blutentnahme zur Berechnung der gemischten venösen Sättigung und bei der Wahl der verwendeten Gleichung, bei der Verwendung von Sedierung oder Anästhesie, die die Hämodynamik beeinflussen kann, bei der Verabreichung hoher Sauerstoffdosen, die die Shunt-Berechnungen verfälschen können, usw. Eine akute Vasodilatator-Provokation wird derzeit in den Leitlinien nicht befürwortet, kann aber Informationen zur Compliance des Lungengefäßsystems liefern ( 1 , 35 ).Während die kurzfristigen Ergebnisse nach Behandlung und Reparatur in Fallberichten und Fallserien vielversprechend waren, bleiben die langfristigen Auswirkungen des Defektverschlusses bei diesen Patienten ungewiss ( 4 , 30 ). Eine zusätzliche Komplikation bei der Behandlung von PAH-CHD-Patienten mit einem Links-Rechts-Shunt ist der Mangel an Daten zur Risikostratifizierung, die die Grundlage für die Entscheidungsfindung in anderen PAH-Kohorten bildet. Frühere Versionen der ESC/ERS-Leitlinien zur pulmonalen Hypertonie schlugen ein erweitertes dreischichtiges Prognosemodell für PAH vor, das die Behandlung leiten sollte, aber für CHD, insbesondere bei Patienten mit Links-Rechts-Shunts, nicht validiert ist ( 29 ). Dieses Modell wurde im Laufe der Jahre verfeinert und es wurden aktualisierte Prognosemodelle auf der Grundlage europäischer und US-amerikanischer Kohorten entwickelt. In den neuesten Leitlinien von 2022 wurde ein vereinfachtes vierschichtiges Modell vorgeschlagen, das 3 klinische Parameter verwendet (WHO-Funktionsklasse, 6-Minuten-Gehstrecke und bran-natriuretisches Peptid); Die beiden wichtigsten Studien, die dieses Modell stützen, schlossen nur sehr wenige Patienten mit angeborener Herzkrankheit ein, die nicht dem Eisenmenger-Modell unterlagen ( 1 , 28 ). Dieses Modell wurde bei Patienten mit angeborener Herzkrankheit (PAH) angewendet, aber nicht formal validiert ( 45 ).5 Vorschlag für ein praktisches VorgehenBei der Erwägung von PAH-Therapien bei Patienten mit PAH-Angeborenen Herzfehlern und Links-Rechts-Shunts können Expertenzentren die folgenden zwei zentralen Fragen stellen:1. Was sind die Therapieziele und welche klinischen Endpunkte werden zur Beurteilung der Wirksamkeit festgelegt?2. Wie sollte man Patienten auswählen, die am wahrscheinlichsten von einer PAH-Therapie profitieren?Die möglichen Ziele der PAH-Therapie in dieser Kohorte sind entweder:- Erreichen der Operabilitätskriterien, d. h. Modifizierung der pulmonalen Hämodynamik, um die empfohlenen Kriterien für eine Reparatur zu erfüllen (als Teil eines Treat-and-Repair-Ansatzes) ( 35 ), oder- eine Verbesserung des Funktionsstatus, der körperlichen Leistungsfähigkeit und möglicherweise der Prognose bewirken.Defekte, die perkutan repariert werden können, sind für einen Behandlungs- und Reparaturansatz attraktiver, da Operationen, die einen kardiopulmonalen Bypass erfordern, erhebliche Risiken bergen (z. B. perioperative pulmonale Hypertoniekrise), selbst bei Patienten unter wirksamer PAH-Therapie. Eine engmaschige perioperative Überwachung ist unerlässlich, um perioperative Komplikationen zu minimieren, mit einer niedrigen Hemmschwelle für eine Eskalation der PAH-Therapien und hämodynamischen Unterstützung, falls erforderlich. Bei einem Patienten, der nun als PAH-CHD nach der Reparatur klassifiziert wird, sind wiederholte invasive hämodynamische Untersuchungen erforderlich, um die Hämodynamik nach der Reparatur festzustellen und über die Intensität der PAH-Therapie zu entscheiden, mit dem Ziel, die Langzeitergebnisse zu optimieren. Unter diesen Umständen ist die Entscheidung, eine oder mehrere Therapien bei einem Patienten mit normalem oder nahezu normalem PVR abzubrechen, schwierig, und große Vorsicht ist geboten, da die PVD höchstwahrscheinlich bestehen bleibt und sich verschlimmern kann.Bei Patienten mit schwererer PVD, aber immer noch mit vorherrschendem Links-Rechts-Shunt (d. h., die die Eisenmenger-Kriterien noch nicht erfüllen), ist eine Behandlungs- und Reparaturstrategie weniger wahrscheinlich erfolgreich; die Reparatur eines Defekts bei einem Patienten, der eine sehr aggressive PAH-Therapie benötigt, um die hämodynamischen Kriterien für die Operabilität zu erreichen, ist möglicherweise nicht ratsam, und der Nutzen der Beseitigung des Links-Rechts-Shunts überwiegt möglicherweise nicht die Risiken des Eingriffs und die Wahrscheinlichkeit einer fortschreitenden schweren PVD nach der Reparatur des Defekts. Es kann jedoch sinnvoll sein, pulmonale Vasodilatatoren zu verwenden, um die ventrikulovaskuläre und Gasaustauschkopplung zu verbessern, das RV-Schlagvolumen zu optimieren und den pulmonalen Blutfluss durch eine bessere Verteilung auf relativ normale V/Q-Einheiten zu verbessern ( 46 ). In diesem Zusammenhang können PAH-Therapien den PVR und die RV-Nachlast senken, obwohl ein Anstieg des Links-Rechts-Shunts und der ventrikulären Volumenüberlastung wahrscheinlich ist. Darüber hinaus ist unklar, wie diese Strategie die natürliche Entwicklung hin zur Eisenmenger-Physiologie beeinflussen (beschleunigen oder verzögern) könnte.Daher ist es sinnvoll, Patienten mit PAH-Angeborenen Herzfehlern und einem Links-Rechts-Shunt in zwei unterschiedliche Gruppen zu unterteilen, vorausgesetzt, es liegt ein großer prä- oder posttrikuspidaler Defekt vor und es liegen keine Erkrankungen des linken Herzens oder damit verbundene Läsionen vor ( Abbildung 2 ):- Der überwiegend reversible PVD-Typ bezieht sich auf Patienten mit einem großen Links-Rechts-Shunt und Anzeichen von typischerweise leichter pulmonaler Hypertonie. Die überwiegende Mehrheit dieser Patienten gilt als operabel, einige fallen jedoch in eine „Grauzone“ der Entscheidungsfindung, die einer sorgfältigen Expertenbeurteilung bedarf. In dieser Gruppe können PAH-Therapien als Teil eines Behandlungs- und Reparaturansatzes eingesetzt werden, obwohl dies nur ausdrücklich für Vorhofseptumdefekte empfohlen wird. Sobald die Reparatur erreicht ist, werden PAH-Therapien typischerweise langfristig fortgesetzt, obwohl je nach Hämodynamik unmittelbar nach dem Eingriff und langfristig eine Anpassung erforderlich sein kann. Patienten, bei denen Behandlung und Reparatur aufgrund eines unzureichenden Abfalls des PVR trotz angemessener Therapien fehlschlagen, haben wahrscheinlich eine überwiegend irreversible Komponente und sollten als solche klassifiziert werden (siehe unten).- Bei der überwiegend irreversiblen PVD oder dem Prä-Eisenmenger -Typ ist der Links-Rechts-Shunt kleiner als für die Größe des Defekts erwartet, aber es liegt weder in Ruhe noch bei Belastung eine Zyanose vor. In dieser Gruppe können PAH-Therapien unter strenger Überwachung in Betracht gezogen werden, um die Hämodynamik und die RV-Funktion zu verbessern, obwohl es keine Beweise dafür gibt. Der mögliche Nutzen von PAH-Therapien muss gegen das Risiko einer Vergrößerung des Links-Rechts-Shunts und eines RV-Versagens aufgrund einer sich verschlechternden Volumenüberlastung abgewogen werden. Bei diesen Patienten ist es unwahrscheinlich, dass die PAH-Therapie zu einem ausreichenden Abfall des PVR führt, der eine Schließung des Defekts ermöglichen würde. Abildung 2
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.