
- Beiträge: 1755
Sidebar
Biomarker, PH
15 Sep 2024 22:24 #2208
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Biomarker, PH wurde erstellt von danny
www.frontiersin.org/journals/cardiovascu....2022.924873/fullEin Überblick über Biomarker für zirkulierende pulmonale arterielle Hypertonie
 Joana Santos-Gomes
1 †
Joana Santos-Gomes
1 †
 Inês Gandra
1 †
Inês Gandra
1 †
 Rui Adão
1
Rui Adão
1
 Frédéric Perros
2,3
Frédéric Perros
2,3
 Carmen Brás-Silva
1,4 *
Carmen Brás-Silva
1,4 *
 Tabelle 1. Aktualisierte klinische Klassifikation der pulmonalen Hypertonie. Charakteristisch für die PAH sind übermäßige pulmonalvaskuläre Umbauten. Dazu gehören eine Mediahypertrophie - ein frühes Ereignis bei PAH, das sogar reversibel ist und bei allen PAH-Untergruppen auftritt -, proliferative und fibrotische Veränderungen der Intima, eine adventive Verdickung und eine Thrombose in situ (
1
–
3
,
8
–
11
). Die Mechanismen, die diesen Transformationen zugrunde liegen, sind nicht vollständig verstanden (
12
). Sie beinhalten mehrere pathophysiologische Prozesse wie Migration und Proliferation von pulmonalarteriellen glatten Muskelzellen (PASMCs) und Endothelzellen (ECs) (
1
,
2
), endotheliale Dysfunktion, endothelial-mesenchymaler Übergang (
13
), verstärkte Proliferation, Migration und Differenzierung von adventitialen pulmonalarteriellen Fibroblasten (
14
), Entzündung (
15
,
16
) und oxidativen Stress (
2
,
3
,
8
,
9
). Insbesondere ECs könnten an der Synthese von Wachstumsfaktoren beteiligt sein, die die Ablagerung nicht-zellulärer Matrix und die Hypertrophie der glatten Muskulatur stimulieren und so zur Bildung plexiformer Läsionen beitragen (
1
,
4
,
10
,
17
). Darüber hinaus können sich nekrotisches und fibrotisches Gewebe sowie Entzündungszellen in der Arterienwand ansammeln, was zu Arteriitis führt (
1
,
2
,
4
). Schließlich kommt es zu Veränderungen in der Produktion vasoaktiver Moleküle, nämlich Stickstoffmonoxid (NO), Prostacycline und Endothelin-1 (ET-1) (
2
,
18
), was zu endothelialer Dysfunktion und Vasokonstriktion beiträgt.Alle diese Veränderungen gipfeln in einer Obstruktion der Lungenarterien mit einem Anstieg des pulmonalvaskulären Widerstandes (PVR), was zu einer Überlastung des rechten Ventrikels und schließlich zu einem Versagen des rechten Ventrikels (RV) führt (
1
,
9
,
19
) – der Hauptursache für Mortalität und Morbidität bei PAH (
8
). Frühe Studien vor der Einführung PAH-spezifischer Therapien deuteten darauf hin, dass die Überlebensdauer der Patienten nach der Diagnose nur 2,8 Jahre betrug (
20
,
21
).Die Patienten weisen Symptome auf, die auf eine Funktionsstörung des rechten Ventrikels hinweisen und unspezifisch sind (
2
,
3
,
18
). Aus diesem Grund verzögert sich die Diagnose oft um mindestens zwei Jahre, und bei vielen Patienten wird die Krankheit erst in fortgeschrittenen Stadien diagnostiziert (
22
). Zur Charakterisierung der PAH verlangen die aktuellen Leitlinien, dass sich die Patienten einer Rechtsherzkatheteruntersuchung unterziehen, wobei ein mittlerer pulmonalarterieller Druck (mPAP) > 20 mmHg, ein pulmonalarterieller Verschlussdruck ≤ 15 mmHg und ein PVR ≥ 3 Wood-Einheiten in Ruhe gemessen werden, sofern keine anderen Ursachen vorliegen (
5
).Bei Diagnose einer PAH muss mit der entsprechenden Behandlung begonnen werden. Die Therapie von PAH-Patienten hat sich in den letzten Jahrzehnten erheblich weiterentwickelt, parallel zu Verbesserungen bei Überlebenschancen und Lebensqualität der Patienten (
21
). Empfohlen wird ein mehrdimensionaler Ansatz, der allgemeine Maßnahmen (überwachte körperliche Rehabilitation, Infektionsprävention und psychosoziale Unterstützung), supportive Therapien (wie Diuretika und zusätzlicher Sauerstoff) und eine zielgerichtete medikamentöse Therapie mit PAH (
17
) umfasst. Spezielle PAH-Therapien zielen auf die drei wichtigsten molekularen Signalwege ab, die im dysfunktionalen Lungenendothel verändert sind. Endothelin-Rezeptor-Antagonisten (ERAs) modulieren den Endothelin-Signalweg (ET); Phosphodiesterase-Typ-5-(PDE-5)-Hemmer und Stimulatoren der löslichen Guanylatcyclase wirken über den NO-Signalweg; und Prostazyklin-Analoga und Prostazyklin-Rezeptor-Agonisten wirken über den Prostanoid-Signalweg. Diese Substanzen wirken hauptsächlich über die Induktion einer pulmonalen Vasodilatation. Die Erstbehandlung erfolgt üblicherweise mit einer Zweifachkombinationstherapie. Die Patienten werden regelmäßig untersucht, um die Krankheit unter Kontrolle zu halten und die Behandlung entsprechend anzupassen (
17
).Die Rechtsherzkatheteruntersuchung ist derzeit der Goldstandard zur Diagnose und Prognose der Erkrankung, insbesondere im Hinblick auf eine Anpassung der Behandlung. Aufgrund ihrer Invasivität kann sie jedoch weder in der klinischen Routine noch in der Patientennachsorge eingesetzt werden. Stattdessen werden sie durch Messungen des systolischen pulmonalarteriellen Drucks (PAP) und eine transthorakale Echokardiographie ersetzt (
23
).Daher ist es wichtig, nicht-invasive Techniken zur Krankheitsüberwachung zu finden. Biomarker können eine wirksame Lösung für die Diagnose und Prognose sowie die Bewertung der Reaktion auf die Therapie sein, solange ihre Erkennung nicht-invasiv, einfach und objektiv ist. Ziel dieser Übersicht ist es, einige der potenziellen Kandidaten als Biomarker für die Diagnose und Prognose von PAH zu erläutern und zu beschreiben.Was ist ein Biomarker?Per Definition ist ein Biomarker „eine Eigenschaft, die objektiv gemessen und als Indikator für normale biologische Prozesse, pathogene Prozesse oder pharmakologische Reaktionen auf einen therapeutischen Eingriff ausgewertet wird“ (
12
,
24
). Kurz gesagt können wir einen Biomarker als eine molekulare Veränderung in Geweben und/oder Körperflüssigkeiten als Folge eines Krankheitsprozesses definieren (
12
). Im Idealfall sollte ein Biomarker klinische Ergebnisse darstellen, d. h. widerspiegeln, wie sich der Patient fühlt und in welchem Stadium der Krankheit er sich befindet. Er sollte auch als Instrument für Diagnose und Prognose sowie als therapeutischer Marker dienen, der Informationen über die Reaktion des Patienten auf eine bestimmte Behandlung liefert (
12
,
25
). Darüber hinaus muss er eine Reihe von Eigenschaften aufweisen, die ihn ideal machen, nämlich hohe Sensitivität und Spezifität, einfache Beschaffung/Erfassung und Messung, vollständige Verfügbarkeit und Nicht-Invasivität. Zudem muss er ein Indikator für Krankheitsaktivität (Risikostratifizierung, Ansprechen auf die Behandlung, Erwartung einer klinischen Verschlechterung) oder ein Behandlungsziel sein (
12
,
25
). Die Suche nach idealen Biomarkern geht weiter, und in Situationen, in denen noch kein idealer Kandidat bekannt ist, wird der bzw. die Biomarker verwendet, die als zugänglicher, billiger und leichter zu messen gelten. Da diese jedoch weniger sensitiv und weniger spezifisch sein können, sollten sie nicht allein als klinisches Entscheidungsinstrument verwendet werden, und es müssen mehrere Faktoren in die Entscheidungsfindung einbezogen werden (
25
).Obwohl noch kein PAH-Biomarker bekannt ist, der durch einen einzigen, einfachen Test nachgewiesen werden kann, gibt es bereits mehrere bekannte und gut definierte Biomarker, die sich in Zukunft als wirksame diagnostische und prognostische Indikatoren erweisen könnten (
23
,
26
). Diese Biomarker können nach dem (patho)physiologischen Mechanismus kategorisiert werden, mit dem sie in Zusammenhang stehen und der die Komplexität dieses Syndroms widerspiegelt: Endothelfunktion, Entzündung, oxidativer Stress, Herzfunktion, Myokardschädigung, Stoffwechsel und Genexpression (
Abbildung 1
). ABILDUNG 1
Tabelle 1. Aktualisierte klinische Klassifikation der pulmonalen Hypertonie. Charakteristisch für die PAH sind übermäßige pulmonalvaskuläre Umbauten. Dazu gehören eine Mediahypertrophie - ein frühes Ereignis bei PAH, das sogar reversibel ist und bei allen PAH-Untergruppen auftritt -, proliferative und fibrotische Veränderungen der Intima, eine adventive Verdickung und eine Thrombose in situ (
1
–
3
,
8
–
11
). Die Mechanismen, die diesen Transformationen zugrunde liegen, sind nicht vollständig verstanden (
12
). Sie beinhalten mehrere pathophysiologische Prozesse wie Migration und Proliferation von pulmonalarteriellen glatten Muskelzellen (PASMCs) und Endothelzellen (ECs) (
1
,
2
), endotheliale Dysfunktion, endothelial-mesenchymaler Übergang (
13
), verstärkte Proliferation, Migration und Differenzierung von adventitialen pulmonalarteriellen Fibroblasten (
14
), Entzündung (
15
,
16
) und oxidativen Stress (
2
,
3
,
8
,
9
). Insbesondere ECs könnten an der Synthese von Wachstumsfaktoren beteiligt sein, die die Ablagerung nicht-zellulärer Matrix und die Hypertrophie der glatten Muskulatur stimulieren und so zur Bildung plexiformer Läsionen beitragen (
1
,
4
,
10
,
17
). Darüber hinaus können sich nekrotisches und fibrotisches Gewebe sowie Entzündungszellen in der Arterienwand ansammeln, was zu Arteriitis führt (
1
,
2
,
4
). Schließlich kommt es zu Veränderungen in der Produktion vasoaktiver Moleküle, nämlich Stickstoffmonoxid (NO), Prostacycline und Endothelin-1 (ET-1) (
2
,
18
), was zu endothelialer Dysfunktion und Vasokonstriktion beiträgt.Alle diese Veränderungen gipfeln in einer Obstruktion der Lungenarterien mit einem Anstieg des pulmonalvaskulären Widerstandes (PVR), was zu einer Überlastung des rechten Ventrikels und schließlich zu einem Versagen des rechten Ventrikels (RV) führt (
1
,
9
,
19
) – der Hauptursache für Mortalität und Morbidität bei PAH (
8
). Frühe Studien vor der Einführung PAH-spezifischer Therapien deuteten darauf hin, dass die Überlebensdauer der Patienten nach der Diagnose nur 2,8 Jahre betrug (
20
,
21
).Die Patienten weisen Symptome auf, die auf eine Funktionsstörung des rechten Ventrikels hinweisen und unspezifisch sind (
2
,
3
,
18
). Aus diesem Grund verzögert sich die Diagnose oft um mindestens zwei Jahre, und bei vielen Patienten wird die Krankheit erst in fortgeschrittenen Stadien diagnostiziert (
22
). Zur Charakterisierung der PAH verlangen die aktuellen Leitlinien, dass sich die Patienten einer Rechtsherzkatheteruntersuchung unterziehen, wobei ein mittlerer pulmonalarterieller Druck (mPAP) > 20 mmHg, ein pulmonalarterieller Verschlussdruck ≤ 15 mmHg und ein PVR ≥ 3 Wood-Einheiten in Ruhe gemessen werden, sofern keine anderen Ursachen vorliegen (
5
).Bei Diagnose einer PAH muss mit der entsprechenden Behandlung begonnen werden. Die Therapie von PAH-Patienten hat sich in den letzten Jahrzehnten erheblich weiterentwickelt, parallel zu Verbesserungen bei Überlebenschancen und Lebensqualität der Patienten (
21
). Empfohlen wird ein mehrdimensionaler Ansatz, der allgemeine Maßnahmen (überwachte körperliche Rehabilitation, Infektionsprävention und psychosoziale Unterstützung), supportive Therapien (wie Diuretika und zusätzlicher Sauerstoff) und eine zielgerichtete medikamentöse Therapie mit PAH (
17
) umfasst. Spezielle PAH-Therapien zielen auf die drei wichtigsten molekularen Signalwege ab, die im dysfunktionalen Lungenendothel verändert sind. Endothelin-Rezeptor-Antagonisten (ERAs) modulieren den Endothelin-Signalweg (ET); Phosphodiesterase-Typ-5-(PDE-5)-Hemmer und Stimulatoren der löslichen Guanylatcyclase wirken über den NO-Signalweg; und Prostazyklin-Analoga und Prostazyklin-Rezeptor-Agonisten wirken über den Prostanoid-Signalweg. Diese Substanzen wirken hauptsächlich über die Induktion einer pulmonalen Vasodilatation. Die Erstbehandlung erfolgt üblicherweise mit einer Zweifachkombinationstherapie. Die Patienten werden regelmäßig untersucht, um die Krankheit unter Kontrolle zu halten und die Behandlung entsprechend anzupassen (
17
).Die Rechtsherzkatheteruntersuchung ist derzeit der Goldstandard zur Diagnose und Prognose der Erkrankung, insbesondere im Hinblick auf eine Anpassung der Behandlung. Aufgrund ihrer Invasivität kann sie jedoch weder in der klinischen Routine noch in der Patientennachsorge eingesetzt werden. Stattdessen werden sie durch Messungen des systolischen pulmonalarteriellen Drucks (PAP) und eine transthorakale Echokardiographie ersetzt (
23
).Daher ist es wichtig, nicht-invasive Techniken zur Krankheitsüberwachung zu finden. Biomarker können eine wirksame Lösung für die Diagnose und Prognose sowie die Bewertung der Reaktion auf die Therapie sein, solange ihre Erkennung nicht-invasiv, einfach und objektiv ist. Ziel dieser Übersicht ist es, einige der potenziellen Kandidaten als Biomarker für die Diagnose und Prognose von PAH zu erläutern und zu beschreiben.Was ist ein Biomarker?Per Definition ist ein Biomarker „eine Eigenschaft, die objektiv gemessen und als Indikator für normale biologische Prozesse, pathogene Prozesse oder pharmakologische Reaktionen auf einen therapeutischen Eingriff ausgewertet wird“ (
12
,
24
). Kurz gesagt können wir einen Biomarker als eine molekulare Veränderung in Geweben und/oder Körperflüssigkeiten als Folge eines Krankheitsprozesses definieren (
12
). Im Idealfall sollte ein Biomarker klinische Ergebnisse darstellen, d. h. widerspiegeln, wie sich der Patient fühlt und in welchem Stadium der Krankheit er sich befindet. Er sollte auch als Instrument für Diagnose und Prognose sowie als therapeutischer Marker dienen, der Informationen über die Reaktion des Patienten auf eine bestimmte Behandlung liefert (
12
,
25
). Darüber hinaus muss er eine Reihe von Eigenschaften aufweisen, die ihn ideal machen, nämlich hohe Sensitivität und Spezifität, einfache Beschaffung/Erfassung und Messung, vollständige Verfügbarkeit und Nicht-Invasivität. Zudem muss er ein Indikator für Krankheitsaktivität (Risikostratifizierung, Ansprechen auf die Behandlung, Erwartung einer klinischen Verschlechterung) oder ein Behandlungsziel sein (
12
,
25
). Die Suche nach idealen Biomarkern geht weiter, und in Situationen, in denen noch kein idealer Kandidat bekannt ist, wird der bzw. die Biomarker verwendet, die als zugänglicher, billiger und leichter zu messen gelten. Da diese jedoch weniger sensitiv und weniger spezifisch sein können, sollten sie nicht allein als klinisches Entscheidungsinstrument verwendet werden, und es müssen mehrere Faktoren in die Entscheidungsfindung einbezogen werden (
25
).Obwohl noch kein PAH-Biomarker bekannt ist, der durch einen einzigen, einfachen Test nachgewiesen werden kann, gibt es bereits mehrere bekannte und gut definierte Biomarker, die sich in Zukunft als wirksame diagnostische und prognostische Indikatoren erweisen könnten (
23
,
26
). Diese Biomarker können nach dem (patho)physiologischen Mechanismus kategorisiert werden, mit dem sie in Zusammenhang stehen und der die Komplexität dieses Syndroms widerspiegelt: Endothelfunktion, Entzündung, oxidativer Stress, Herzfunktion, Myokardschädigung, Stoffwechsel und Genexpression (
Abbildung 1
). ABILDUNG 1
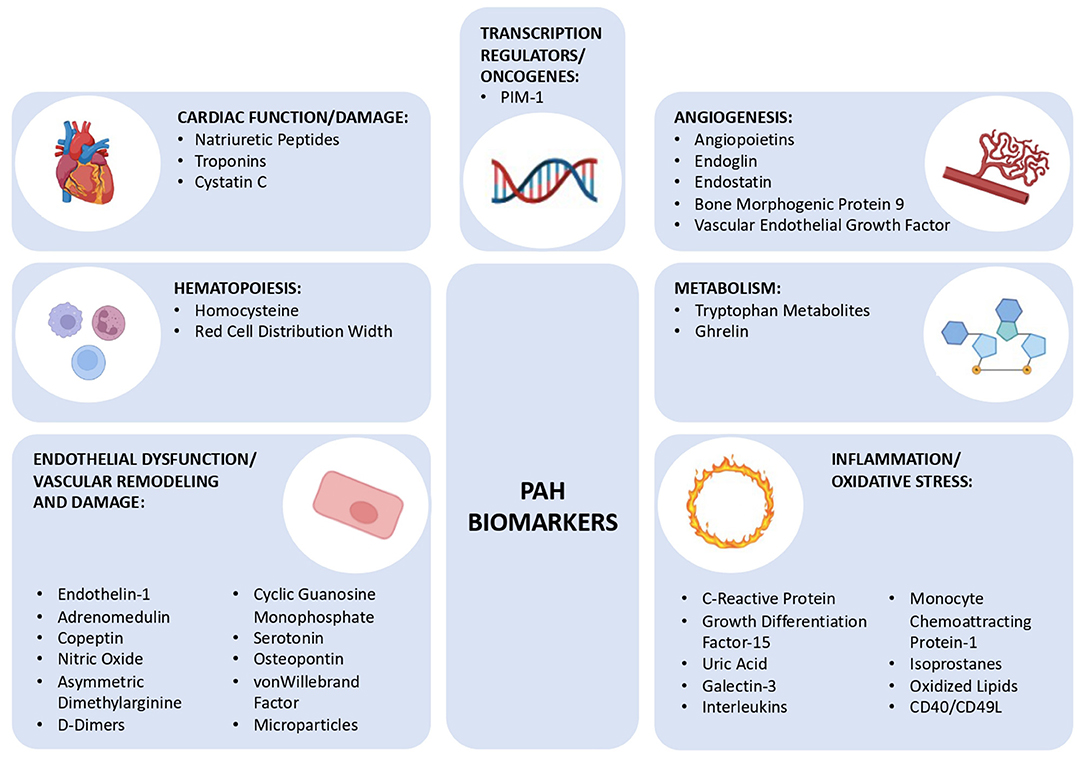 Abbildung 1. Schematische Darstellung der verschiedenen Biomarkergruppen für pulmonale arterielle Hypertonie, die in dieser Übersicht besprochen werden, unterteilt in 7 Gruppen: Herzfunktion/-schäden, Hämatopoese, endotheliale Dysfunktion/vaskuläre Umgestaltung und Schäden, Angiogenese, Stoffwechsel, Entzündung/oxidativer Stress und Transkriptionsregulatoren/Onkogene. PIM-1, Moloney Murine Leukemia Provirus Integration Site. Biomarker der Herzfunktion/-schädigungNatriuretische PeptideNatriuretische Peptide sind eine Familie genetisch unterschiedlicher Hormone mit ähnlicher Molekularstruktur (
12
). Zu dieser Familie gehören das atriale natriuretische Peptid (ANP), das brachiale natriuretische Peptid (BNP), das C-Typ-natriuretische Peptid (
3
,
27
) und das Dendroaspis-natriuretische Peptid, ein D-Typ-natriuretisches Peptid, die jeweils spezifische Funktionen haben (
27
). Sie spielen eine grundlegende Rolle bei der kardialen Homöostase (
28 ), da sie durch ihre diuretische, natriuretische, vasodilatatorische (
3
,
29
) und kaliuretische (
27
) Wirkung an der Regulierung des Blutvolumens und des Blutdrucks beteiligt sind. Darüber hinaus hemmen sie das Renin-Angiotensin-Aldosteron-System (
27
,
29
) und regulieren die Proliferation endokriner Zellen (
27
). Diese Hormone werden im Wesentlichen vom Herzen, den Nieren und dem Gehirn ausgeschüttet (
3
).ANP und BNP sind die Haupthormone des natriuretischen Peptidsystems (
12
,
30
). Beide werden von Herzmuskelzellen freigesetzt. ANP wird hauptsächlich vom Vorhofgewebe freigesetzt, während BNP vom Ventrikelgewebe freigesetzt wird. Beide werden als Reaktion auf erhöhten Herzdruck und Volumenüberlastung ausgeschüttet (
31
). BNP wird vom Ventrikelgewebe ausgeschüttet und reagiert empfindlicher auf Ventrikelerkrankungen als ANP (
12
).BNP ist ein Transkriptionsprodukt des NPPB-Gens , das zunächst einen Vorläufer mit 134 Aminosäuren (aa) bildet, PräproBNP, das enzymatisch gespalten wird, wobei proBNP entsteht (
3
,
30
). ProBNP wird in zwei verschiedene Fragmente gespalten: ein aktives Peptid, reifes 32 aa BNP und ein inaktives N-terminales Fragment, das N-terminale Prohormon (NT-proBNP) (
3
,
27
,
30
). BNP wirkt, indem es an Rezeptor A bindet, der hauptsächlich in Nieren, Nebennieren, Lunge, terminalem Ileum, Aorta und Fettgewebe exprimiert wird (
3
). Die Aktivierung dieses Rezeptors induziert einen Anstieg des zyklischen Guanosinmonophosphats (cGMP), was eine vasodilatatorische und natriuretische Reaktion auslöst und Aldosteron hemmt (
3
). Obwohl die Funktion von NT-proBNP noch nicht geklärt ist (
3
), wird es über die Nieren ausgeschieden, weshalb seine Plasmawerte bei Veränderungen der Nierenfunktion signifikant ansteigen. Dies ist bei BNP nicht der Fall (
30
). Reifes BNP hat eine kurze Plasmahalbwertszeit (etwa 22 Minuten), während NT-proBNP eine Halbwertszeit von 2 Stunden hat (
30
) und daher stabiler ist (
3
,
12
) und leichter messbar ist (
12
). Die Werte von kardialem und zirkulierendem BNP steigen als Reaktion auf Hypertrophie und/oder ventrikuläre Überlastung signifikant an, was zeigt, dass BNP ein ausgezeichneter Marker für ventrikuläre Dysfunktion ist (
28
).Zur Pathophysiologie der PH: BNP ist bei verschiedenen Formen der PAH erhöht, darunter idiopathische PAH (IPAH) (
32
), PAH in Verbindung mit Bindegewebserkrankungen (CTD-PAH) (
33
), angeborene systemisch-pulmonale Shunts (
34
), PH in Verbindung mit Lungenerkrankungen (
35
), PH und chronisch obstruktiver Lungenerkrankung (COPD) (
28
), chronisch thromboembolische PH (CTEPH) (
29
) und PH in Verbindung mit akuter Lungenembolie (
36
). In mehreren Studien korrelieren BNP-Spiegel eng mit der Funktionsklasse der New York Heart Association (NYHA), dem 6-Minuten-Gehstrecke (6MWD)-Test und hämodynamischen Parametern (
3
,
29
,
32
,
37
). Nagaya et al. es zeigte sich auch, dass BNP-Spiegel positiv mit mPAP und umgekehrt mit dem Herzzeitvolumen korrelieren, wobei eine starke Korrelation mit dem totalen Lungenwiderstand besteht (
38
). NT-proBNP ist auch bei verschiedenen Formen der PAH erhöht, wie etwa bei IPAH (
28
) und bei systemischer Sklerose-assoziierter PAH (SSc-PAH) (
39
). Im letzteren Fall korreliert NT-proBNP mit mPAP, PVR, mittlerem rechtsatrialen Druck (mRAP) und Herzindex (
3
) und kann wirksam als Prädiktor des Überlebens bei PAH dienen (
23
). ANP-Spiegel sind auch bei IPAH (
40
) und CTEPH erhöht. Wie BNP korrelieren ANP-Spiegel positiv mit mPAP und umgekehrt mit dem Herzzeitvolumen (
38
). Allerdings hat ANP bei Menschen eine kurze Halbwertszeit von etwa 2 Minuten (
41
,
42
) - während BNP eine viel längere Halbwertszeit hat (
43
), was BNP zu einem besseren Kandidaten als Biomarker macht (
3
).Somit sind BNP und NT-proBNP sensitive und spezifische Biomarker für die Risikostratifizierung bei PAH (sie sind bislang die einzigen Biomarker, die in den aktuellen Leitlinien enthalten sind) (
23
). Sie sind zudem sensitive Marker für RV-Dysfunktionen und die Wirksamkeit der Behandlung (
23
,
26
,
28
). Zudem besitzt BNP als pulmonaler Vasodilatator und antihypertropher Wirkstoff das therapeutische Potenzial, pulmonalvaskuläre Umbauprozesse zu lindern (
28
). Dabei müssen jedoch immer bestimmte Faktoren berücksichtigt werden, nämlich Geschlecht und Alter des Patienten sowie das Vorliegen einer Linksherzerkrankung, Nierenfunktionsstörungen und Fettleibigkeit (
12
,
26
). In Fällen einer Linksherzerkrankung sollten BNP-Spiegel nicht zur Vorhersage der Diagnose oder Prognose von PAH herangezogen werden (
30
).TroponinKardiale Troponine (cTn) sind eine Gruppe von 3 Proteinen – Troponin C, Troponin I (cTnI) und Troponin T (cTnT) –, deren Hauptfunktion darin besteht, die dünnen Aktinfilamente des Herzmuskels zu regulieren. Der Troponinspiegel hängt eng mit der Schädigung des Myokards zusammen, da das Troponin beim Zerreißen der Kardiomyozytenmembranen ins periphere Blut freigesetzt wird. Daher können sie mittels hochsensibler Tests im Plasma nachgewiesen werden (
12
,
26
). Die Messung von cTnT und cTnI ist von entscheidender Bedeutung für die Diagnose und Prognose von Patienten mit akutem Koronarsyndrom sowie für alle Pathologien, die mit Myokardläsionen einhergehen und erhöhte Werte kardialer Troponine aufweisen, wie z. B. bei Myokarditis (
12
).In der Pathophysiologie der PH: Mehrere Studien haben cTnT mit der schlechten Prognose von PH in Verbindung gebracht. Torbicki et al. zeigten, dass bei Patienten mit PAH und CTEPH die cTnT-Werte erhöht sind, was wahrscheinlich durch die Schädigung des RV-Myokards erklärt werden kann. In dieser Studie hatten jedoch nur 14 % der Patienten (8 von 56 Patienten) erhöhte cTnT-Werte, da der Test zur Messung der cTnT-Werte nur die Erkennung von Werten >0,01 ng/ml ermöglicht. Beim Vergleich von cTnT(+) - und cTnT(-)-Patienten zeigte sich dennoch, dass sie eine ähnliche pulmonale Hämodynamik hatten, aber cTnT(+)-Patienten eine höhere Herzfrequenz, eine geringere gemischtvenöse Sauerstoffsättigung (SvO 2 ), höhere NT-proBNP-Serumwerte und eine geringere Belastungsresistenz (gemessen mit 6MWD) (
44
). Darüber hinaus wurde bei Patienten unter PH-Behandlung festgestellt, dass die cTnT-Spiegel sinken und sogar unter die Nachweisgrenze geraten; im Gegensatz dazu steigen die Werte mit Fortschreiten der Erkrankung an (
44
).Neuere Studien bieten Tests mit höherer Sensitivität gegenüber cTn, aber deutlich niedrigeren Nachweisgrenzen. Heresi et al. zeigten unter Verwendung eines Immunanalysators mit einer Nachweisgrenze von < 0,008 ng/ml, dass bei 25 % der Patienten mit PAH eine Erhöhung des cTnI-Werts festgestellt werden konnte. Im Vergleich zu cTnI(+)-Patienten hatten cTnI(+)-Patienten eine höhere Funktionsklasse, einen größeren rechten Vorhofbereich, eine niedrigere 6MWD und höhere BNP- und C-reaktive Protein- (CRP-) Werte. Zudem war die Überlebensrate von cTnI(+)-Patienten signifikant niedriger als die von cTnI(-)-Patienten (44 % vs. 85 %) (
45
). Filusch et al. untersuchten die cTnT-Werte bei Patienten mit PAH und verglichen den konventionellen Test mit dem hochsensitiven cTnT-Test (hsTnT) mit einer Nachweisgrenze von weniger als 2 pg/ml. Bei 90,9 % der Patienten war cTnT mit dem hsTnT-Test nachweisbar, im Vergleich zu 30,9 % mit dem herkömmlichen Test. Außerdem waren die Messungen des hsTnT-Tests signifikant mit einer systolischen RV-Dysfunktion und einer beeinträchtigten 6MWD verbunden. Darüber hinaus sagte hsTnT eine Funktionsklasse II oder höher der Weltgesundheitsorganisation (WHO) besser voraus als NT-proBNP und sagte den Tod genauso effektiv voraus wie NT-proBNP (
46
). In einer anderen Studie waren mit dem neuen hsTnI-Test cTnI-Werte bei 95 % der Patienten mit PH nachweisbar, einschließlich PAH-Patienten. Höhere cTnI-Werte sind mit höheren BNP-Werten, einer niedrigeren 6MWD, schwerwiegenderen hämodynamischen Anomalien und Anomalien in der kardialen MRT-Bildgebung verbunden (
47
).Obwohl cTn auch mit einigen Markern auf der linken Seite des Herzens in Zusammenhang stehen (
47
), was ein Störfaktor sein kann, wiesen Studien darauf hin, dass sie als Indikatoren für die Schwere der Erkrankung verwendet werden können. Darüber hinaus liefert die Verwendung der neuen hsTn-Tests neue prognostische Informationen, und diese Tests haben das Potenzial, mehr Patienten mit höherem Risiko zu erkennen und so die Risikostratifizierung zu erleichtern. Dennoch gibt es Störfaktoren, die berücksichtigt werden müssen, nämlich das Vorhandensein einer gleichzeitigen Erkrankung der linken Herzhälfte oder eines Nierenversagens (
12
,
26
).Cystatin CCystatin C (CysC) ist ein 13 kDa großes, nicht-glykosyliertes Protein (
48
,
49
), wird von allen untersuchten kernhaltigen Zellen mit konstanter Rate produziert (
50
) und ist ein Mitglied der Cystatin-Superfamilie, die Inhibitoren von Cysteinproteinasen umfasst (
51
). In den letzten Jahren hat CysC aufgrund seiner freien Filtration im Glomerulus, seiner vollständigen Reabsorption und Katabolisierung im proximalen Tubulus und seines Fehlens einer tubulären Sekretion an Bedeutung gewonnen. Man nimmt an, dass die Plasmakonzentration von CysC fast vollständig von der glomerulären Filtrationsrate abhängt, und macht es daher zu einem idealen Marker der Nierenfunktion (
48
,
49
). Außerdem ist es sensitiver als Serumkreatin, da es geringere Verringerungen der glomerulären Filtrationsrate erkennt (
48
,
49
). Mehrere Studien haben gezeigt, dass CysC auch als kardiovaskulärer Risikomarker fungieren kann, da es Linksherzinsuffizienz (HF) und kardiovaskuläre Mortalität im Allgemeinen vorhersagt (
48
,
49
).In der PH-Pathophysiologie: Fenster et al. zeigten, dass Patienten mit PAH abnorm hohe CysC-Serumspiegel haben und dass diese Spiegel mit der RV-Funktion korrelieren (
52
). Diese Studien zeigen, dass der systolische RV-Druck bei PAH-Patienten stark erhöht ist und positiv mit CysC-Serumspiegeln korreliert. Darüber hinaus korrelieren RV-enddiastolisches Volumen, RV-endsystolisches Volumen, Masseindex, Dehnung und Dehnungsrate positiv mit CysC-Spiegeln und die RV-Ejektionsfraktion negativ (
52
). Somit kann CysC ein sensitiver Biomarker zur Beurteilung von PAH sein und hat gegenüber den Standardbiomarkern BNP und NT-proBNT zusätzliche Vorteile, da es unabhängig von Muskelmasse, Alter und Geschlecht ist (
50
).Biomarker der HämatopoeseHomocysteinHomocystein ist ein schwefelhaltiges Zwischenprodukt des normalen Stoffwechsels von Methionin, einer essentiellen Aminosäure, die im Wesentlichen aus tierischem Eiweiß gewonnen wird (
53
). Homocystein muss über einen Weg wiederverwertet werden, der Folsäure und die Vitamine B6 und B12 erfordert . Veränderungen in diesem Stoffwechselweg, wie beispielsweise ein Mangel an Vitamin B6 und B12 , genetische Defekte oder Polymorphismen in den wichtigsten Enzymen dieses Stoffwechselwegs, können zu einem Anstieg des Homocysteinspiegels führen, der schädlich sein kann (
53
). Homocystein ist ein Hemmer der Dimethylarginin-Dimethylaminohydrolase, eines Enzyms, das asymmetrisches Dimethylarginin (ADMA) metabolisiert; ADMA ist ein endogener Hemmer des NO-Synthase(NOS)-Stoffwechselwegs. Daher führt ein Anstieg des Homocysteinspiegels, die sogenannte Hyperhomocysteinämie, zu einer verringerten Bioverfügbarkeit von NO. Man geht davon aus, dass dieser NO-Abnahme darauf zurückzuführen ist, dass Homocystein die Aktivität der Dimethylarginin-Dimethylaminohydrolase hemmt und so die Ansammlung von ADMA fördert und infolgedessen die NO-Produktion durch die endothelialen Zellen verringert (
54
). Darüber hinaus wird angenommen, dass Homocystein am oxidativen Abbau von NO beteiligt ist (
54
,
55
). Da Homocystein im Wesentlichen die endothelialen Zellen, die Blutgerinnung und die Thrombozytenfunktion beeinflusst, erklärt sich, warum die endotheliale Vasodilatatorfunktion bei Personen mit Hyperhomocysteinämie beeinträchtigt und mit einer Thrombozytenfunktionsstörung einhergeht, die die Blutgerinnung erleichtert und somit das Risiko für Herz-Kreislauf-Erkrankungen erhöht und die Entwicklung von Gefäßerkrankungen beschleunigt (
54
,
55
).In der Pathophysiologie der PH zeigen Studien, dass die Gesamthomocysteinwerte im Plasma – einschließlich Homocystein und seiner oxidierten Derivate – gemischtes Homocystein-Cystein-Disulfid und proteingebundenes Homocystein – bei Patienten mit PAH höher sind als bei gesunden Kontrollpersonen (
53
). Darüber hinaus zeigten Sanli et al., dass die Homocysteinwerte bei Patienten mit PAH in Verbindung mit angeborenen Herzfehlern (CHD-PAH) höher sind, ebenso wie die ADMA-Werte, sie korrelierten jedoch nicht mit hämodynamischen Faktoren (
56
). Beide Studien legen also nahe, dass Homocystein ein wichtiger Faktor bei der Entwicklung von PAH sein könnte, was sich durch die Endothelschäden durch Hyperhomocysteinämie gut begründen lässt. Es sollten jedoch weitere Studien durchgeführt werden, da die Stichprobengröße in beiden vorhandenen Studien relativ gering ist (
56
).Verteilungsbreite der roten BlutkörperchenDie Erythrozytenverteilungsbreite (RDW) ist ein Parameter, der die Schwankungen in der Größe der zirkulierenden roten Blutkörperchen widerspiegelt; sie wird routinemäßig bei einem großen Blutbild gemessen und ist von entscheidender Bedeutung für die Differentialdiagnose einer Anämie (
57
–
59
). Mehrere Studien haben gezeigt, dass RDW-Werte bei der Vorhersage bösartiger Tumoren hilfreich sein können (
60
,
61
). Die RDW ist mit verschiedenen pathophysiologischen Mechanismen verbunden, wie Entzündungen, Eisenstoffwechsel, Nierenfunktionsstörungen, Ernährungszustand und oxidativem Stress, die zu einer Verringerung der erythropoetischen Leistung führen (
62
). Erhöhte RDW-Werte stehen in engem Zusammenhang mit einer beeinträchtigten Erythropoese oder dem Abbau von Erythrozyten (
63
). Die RDW hat sich als vielversprechender Prädiktor des klinischen Ausgangs von Nierenerkrankungen, Herz-Kreislauf-Erkrankungen und Lungenerkrankungen (
57
–
59
) wie HF, PH verschiedener Ätiologien (
58
), akutem Myokardinfarkt, ambulant erworbener Pneumonie, Lungenembolie und COPD erwiesen; zudem erwies sie sich als Prädiktor der Mortalität bei Patienten mit COPD und PAH (
64
).In der Pathophysiologie der PH: Ulrich et al. zeigten, dass RDW ein Faktor ist, der mit dem Überleben bei PAH korreliert; und dass bei Patienten mit PAH üblicherweise Eisenmangel auftritt (
65
,
66
). Die RDW-Werte steigen mit sinkenden Eisenwerten, da die verfügbaren Eisenwerte des Körpers nicht dem Eisenbedarf aus der Synthese roter Blutkörperchen entsprechen, was zu unterschiedlichen Größen der roten Blutkörperchen führt (
67
). Rhodes et al. zeigten, dass RDW-Werte mit der WHO-Funktionsklasse und der 6MWD korrelieren (
68
); sie zeigten auch, dass RDW als unabhängiger Prädiktor des Überlebens fungieren kann, auch wenn es in Kombination mit 6MWD, NT-proBNP und anderen klinischen Indizes gemessen wird (
68
).Zusammenfassend lässt sich sagen, dass RDW eng mit der Schwere der Erkrankung zusammenhängt und zur Vorhersage des Überlebens von Patienten mit IPAH verwendet werden kann (
67
,
68
). Daher sollte die Verwendung von RDW als PAH-Biomarker für neue Ansätze mit mehreren Biomarkern zur PAH-Stratifizierung in Betracht gezogen werden, da die RDW-Werte in Kombination mit den NT-proBNP-Werten eine bessere Erkennung von Hochrisikofällen zeigten als die Verwendung von NT-proBNP allein (
68
).Biomarker für endotheliale Dysfunktion und/oder Gefäßumbau und -schädigungEndothelin-1Das ET-System besteht aus drei ET-Isopeptiden (ET-1, ET-2 und ET-3), die Peptidasen mit unterschiedlichen Isoformen aktivieren, und zwei G-Protein-gekoppelten Rezeptoren – dem ET-Typ-A-Rezeptor (ETA) und dem ET-Typ-B-Rezeptor (ETB) (
69
). ETs sind Isopeptide mit 21 Aminosäuren und weisen eine hohe Homologie und Ähnlichkeit zueinander auf. Sie werden hauptsächlich in ECs exprimiert; sie werden jedoch auch in Herzmyozyten, Lungenepithel, glomerulären Nierenzellen, Mesangialzellen, glatten Muskelzellen (SMCs), Leukozyten, Makrophagen (
7
,
70
) und Fibroblasten (
12
) exprimiert. ETs gelten als die wirksamsten endogenen Vasokonstriktoren (
69
), darüber hinaus sind sie aber auch multifunktionelle Peptide mit zytokin- oder hormonähnlicher Aktivität (
71
).ET-1 ist die am stärksten exprimierte Form im Herzkreislaufsystem (
7
) und in den Lungengefäßen (
12
); es bindet an beide Rezeptoren. ETA und ETB sind in verschiedenen Geweben und Zellen verteilt, ihre Expression ist jedoch variabel; ETA wird vorwiegend auf vaskulären glatten Muskelzellen (VSMCs) (
71
) und Myozyten (
7
) exprimiert, während ETB vorwiegend auf ECs (ETB1-Rezeptoren) und VSMCs (ETB2-Rezeptoren) (
7
) lokalisiert ist. Die Aktivierung von ETA vermittelt vor allem Vasokonstriktion sowie Zellproliferation (
69
). Im Gegensatz dazu fördert die Aktivierung von ETB in ECs eine indirekte vasodilatatorische Wirkung, hemmt die Proliferation von Myokard- und Gefäßgewebe und reguliert den Nierenblutdruck. Auf der anderen Seite spielen ETB2-Rezeptoren eine gewisse vasokonstriktorische Rolle, ihre wichtigste Wirkung ist jedoch die ET-1-Clearance (
69
). Die Vasokonstriktion wird durch die Aktivierung von Phospholipase C, einen Anstieg von Inositoltriphosphat und Diacylglycerol und einen anschließenden Anstieg von intrazellulärem Calcium vermittelt, was die Zellkontraktion fördert. Andererseits stimuliert die durch die Aktivierung des endothelialen Rezeptors ETB1 vermittelte Vasodilatation die Freisetzung von NO und Prostacyclinen, was zur Entspannung der Gefäßwand führt (
7
). Die ET-1-Clearance erfolgt durch Internalisierung des Rezeptors, an den ET-1 gebunden hat; die Lunge eliminiert etwa 50 % des zirkulierenden ET-1 (
7
).ET-1 induziert eine intensive und anhaltende Vasokonstriktion der Lungenarterien und -venen, selbst wenn es in niedriger Konzentration vorhanden ist, und kann auch die Proliferation von Lungenfibroblasten stimulieren. Auf kardialer Ebene ist ET-1 an der Steigerung der myokardialen Kontraktilität und der Herzfrequenz beteiligt (positiv inotroper bzw. chronotroper Effekt) und stimuliert darüber hinaus die Produktion von Zytokinen, Wachstumsfaktoren und Matrixproteinen in anderen Geweben (
7
).In der Pathophysiologie der PH weisen die meisten Patienten erhöhte ET-1-Werte auf (
72
). ET-1 wird als Mediator für erhöhten Gefäßtonus und Gefäßumbau angesehen (
73
). Bei PAH kommt es zu einer deutlichen Erhöhung der ET-1-Expression in den Lungengefäßen (
73
), einschließlich der für die Erkrankung charakteristischen plexiformen Läsionen (
7
). Zudem sind die ET-1-Plasmaspiegel erhöht und stehen in enger Korrelation mit RAP und der Sauerstoffsättigung der Lungenarterie (
5
,
74
), PVR und 6MWD (
75
). Eine für PAH charakteristische Endothelschädigung verstärkt die konstriktive Wirkung von ET-1, verursacht eine Dysregulation im ET-System (
7
) und verringert die Fähigkeit des Endothels, Vasodilatatoren freizusetzen (
72
). Diese Dysregulation und Überexpression von ET-1 fördern also einen Anstieg des PVR (
7
), was teilweise auf den Mangel an Vasodilatatoren (
72
) und auf eine abnorme pulmonale Gefäßumgestaltung (
19
) zurückzuführen ist. Hohe ET-1-Konzentrationen sind auch mit einer Entzündungsreaktion und verstärkter Fibrose assoziiert (
1
). Der Anstieg der ET-1-Plasmakonzentrationen bei PAH kann auf eine erhöhte ET-1-Freisetzung oder eine Verringerung der ET-1-Clearance durch das Lungengefäßsystem oder sogar auf eine Kombination beider Faktoren zurückzuführen sein (
1
,
72
).Darüber hinaus ist die Hemmung der ET-Rezeptoren durch ERAs in der Behandlung von PAH effektiv, da sie PAP verringert und die vaskuläre Umgestaltung hemmt. Bosentan zeigte Verbesserungen der hämodynamischen Parameter, der 6MWD und der WHO-Funktionsklasse bei Patienten mit PAH (
76
); Sitaxsentan zeigte Verbesserungen bei 6MWD, WHO-Funktionsklasse, PVR und Herzindex (
77
). Ambrisentan verbessert die körperliche Belastbarkeit, die WHO-Funktionsklasse, die hämodynamischen Parameter und die Sterblichkeit bei PAH-Patienten; zudem ist es mit einem geringen Risiko von Aminotransferaseanomalien verbunden (
78
,
79
). Darüber hinaus zeigte die AMBITION-Studie, dass eine Zweifachkombinationstherapie mit Ambrisentan und Tadalafil, einem PDE-5-Hemmer, das Risiko eines PAH-bedingten Krankenhausaufenthalts im Vergleich zu einer Monotherapie um 63 % senkte (
80
). Macitentan verbessert mPAP, RAP, PVR, Herzindex und die NT-proBNP-Werte (
81
).Daher spielt das ET-1-System eine grundlegende Rolle in der Pathologie der PAH und kann sogar als prognostischer Marker der Krankheit verwendet werden. Die Verwendung von ET-1 als Marker ist jedoch mit einigen Einschränkungen verbunden, die berücksichtigt werden müssen: Da es sich im Wesentlichen über Gefäßstrukturen ausbreitet, spiegeln seine Plasmaspiegel die ET-1-Konzentration im Gewebe nicht genau wider (
12
,
72
). Außerdem müssen demografische Merkmale wie Ethnizität, Geschlecht und Alter immer berücksichtigt werden, da die ET-1-Plasmaspiegel bei Menschen afrikanischer Abstammung, Männern und höherem Alter höher sind und somit potenzielle Störfaktoren darstellen (
82
).AdrenomedullinAdrenomedullin (ADM) ist ein 52 Aminosäuren langes Peptidhormon, das mit einer langanhaltenden pulmonal-vasodilatatorischen Wirkung in Verbindung gebracht wird (
83
,
84
). ADM wurde erstmals 1993 aus menschlichem Phäochromozytom isoliert (
83
,
84
) und später als zirkulierendes Hormon entdeckt (
12
). ADM wird von endothelialen Zellen (ECs) und endothelialen Zellmembranen (VSMCs) produziert und diffundiert zwischen Blut und Interstitium (
83
,
85
). Obwohl es im ganzen Körper Rezeptoren und Bindungsstellen gibt, ist die Rezeptordichte in den Herz-Kreislauf- und Lungengeweben höher, weshalb es hauptsächlich in diesen beiden Systemen wirkt (
83
,
85
). Neben seiner gefäßerweiternden Wirkung hat ADM auch diuretische und natriuretische Wirkungen, hemmt das Renin-Angiotensin-Aldosteron-System und ist an der Angiogenese und der Regulierung von Entzündungen beteiligt (
12
). Aufgrund seiner gefäßerweiternden Wirkung sowie seiner natriuretischen und diuretischen Wirkung ist es verständlich, dass ADM an der Regulierung der Körperflüssigkeit und damit an der Herzhomöostase beteiligt ist (
85
). Außerdem hat es die Fähigkeit, als Regulator des pulmonalen Gefäßtonus und der Gefäßumgestaltung zu wirken (
83
). Frühere Studien zeigten, dass die Plasmaspiegel von ADM bei Patienten mit Bluthochdruck und Herzinsuffizienz erhöht sind.In der Pathophysiologie der PH sind die Plasmaspiegel von ADM bei Patienten mit PAH erhöht und steigen proportional zum Schweregrad der PH an (
83
,
85
). Zudem korrelieren ADM-Spiegel mit klinischen Parametern, einschließlich mRAP-, 6MWD- und NT-proBNP-Spiegeln, mit ESC/ERS- und REVEAL-Risikoscores und können das Gesamtüberleben der Patienten widerspiegeln (
86
). Studien zeigten auch die Bedeutung von ADM als therapeutisches Ziel für PAH – die Gabe von exogenem ADM führte bei Patienten mit PAH zu signifikanten hämodynamischen Verbesserungen (Anstieg des Herzindex und Rückgang des PVR) (
83
); somit wirkt es als krankheitsregulierendes Hormon bei PAH und zusätzlich als alternativer Prognose- und Schweregradmarker (
86
).CopeptinCopeptin ist ein 39 Aminosäuren langes Glykopeptid und ist vor allem als Arginin-Vasopressin (AVP)-assoziiertes Glykopeptid bekannt (
87
). Copeptin wird, wie auch AVP und Neurophysin II, von einem Vorläufer, dem Prä-Pro-Vasopressin, abgeleitet (
87
,
88
). Diese drei Peptide werden stöchiometrisch von der Hypophyse sezerniert, und es ist möglich, Copeptinwerte als Indikator für AVP-Werte zu verwenden (
87
,
88
). AVP wird im Hypothalamus produziert und von der Hypophyse als Reaktion auf hämodynamische und/oder osmotische Reize sezerniert (
87
). AVP bindet an zwei Rezeptortypen: Vasopressin1, das die arterioläre Vasokonstriktion vermittelt (
87
,
88
), und Vasopressin2, das antidiuretisch wirkt (
87
), indem es die Wasserrückresorption durch die Induktion von Aquaporinen in den Sammelrohren der Niere fördert (
88
). AVP hat jedoch eine kurze Plasmahalbwertszeit (
89
), und da es instabil ist, ist das zirkulierende AVP hauptsächlich an Blutplättchen gebunden (
88
) und daher nicht messbar. Copeptin ist ein Prognosemarker für verschiedene kardiovaskuläre Pathologien (
12
,
87
,
88
).In der PH-Pathophysiologie sind die Konzentrationen des zirkulierenden Copeptins bei PAH-Patienten erhöht und korrelieren positiv mit der NYHA-Klasse und negativ mit der 6MWD (
88
). Darüber hinaus wurde festgestellt, dass der Anstieg des Plasmavolumens und niedrige Natriumkonzentrationen im P
Abbildung 1. Schematische Darstellung der verschiedenen Biomarkergruppen für pulmonale arterielle Hypertonie, die in dieser Übersicht besprochen werden, unterteilt in 7 Gruppen: Herzfunktion/-schäden, Hämatopoese, endotheliale Dysfunktion/vaskuläre Umgestaltung und Schäden, Angiogenese, Stoffwechsel, Entzündung/oxidativer Stress und Transkriptionsregulatoren/Onkogene. PIM-1, Moloney Murine Leukemia Provirus Integration Site. Biomarker der Herzfunktion/-schädigungNatriuretische PeptideNatriuretische Peptide sind eine Familie genetisch unterschiedlicher Hormone mit ähnlicher Molekularstruktur (
12
). Zu dieser Familie gehören das atriale natriuretische Peptid (ANP), das brachiale natriuretische Peptid (BNP), das C-Typ-natriuretische Peptid (
3
,
27
) und das Dendroaspis-natriuretische Peptid, ein D-Typ-natriuretisches Peptid, die jeweils spezifische Funktionen haben (
27
). Sie spielen eine grundlegende Rolle bei der kardialen Homöostase (
28 ), da sie durch ihre diuretische, natriuretische, vasodilatatorische (
3
,
29
) und kaliuretische (
27
) Wirkung an der Regulierung des Blutvolumens und des Blutdrucks beteiligt sind. Darüber hinaus hemmen sie das Renin-Angiotensin-Aldosteron-System (
27
,
29
) und regulieren die Proliferation endokriner Zellen (
27
). Diese Hormone werden im Wesentlichen vom Herzen, den Nieren und dem Gehirn ausgeschüttet (
3
).ANP und BNP sind die Haupthormone des natriuretischen Peptidsystems (
12
,
30
). Beide werden von Herzmuskelzellen freigesetzt. ANP wird hauptsächlich vom Vorhofgewebe freigesetzt, während BNP vom Ventrikelgewebe freigesetzt wird. Beide werden als Reaktion auf erhöhten Herzdruck und Volumenüberlastung ausgeschüttet (
31
). BNP wird vom Ventrikelgewebe ausgeschüttet und reagiert empfindlicher auf Ventrikelerkrankungen als ANP (
12
).BNP ist ein Transkriptionsprodukt des NPPB-Gens , das zunächst einen Vorläufer mit 134 Aminosäuren (aa) bildet, PräproBNP, das enzymatisch gespalten wird, wobei proBNP entsteht (
3
,
30
). ProBNP wird in zwei verschiedene Fragmente gespalten: ein aktives Peptid, reifes 32 aa BNP und ein inaktives N-terminales Fragment, das N-terminale Prohormon (NT-proBNP) (
3
,
27
,
30
). BNP wirkt, indem es an Rezeptor A bindet, der hauptsächlich in Nieren, Nebennieren, Lunge, terminalem Ileum, Aorta und Fettgewebe exprimiert wird (
3
). Die Aktivierung dieses Rezeptors induziert einen Anstieg des zyklischen Guanosinmonophosphats (cGMP), was eine vasodilatatorische und natriuretische Reaktion auslöst und Aldosteron hemmt (
3
). Obwohl die Funktion von NT-proBNP noch nicht geklärt ist (
3
), wird es über die Nieren ausgeschieden, weshalb seine Plasmawerte bei Veränderungen der Nierenfunktion signifikant ansteigen. Dies ist bei BNP nicht der Fall (
30
). Reifes BNP hat eine kurze Plasmahalbwertszeit (etwa 22 Minuten), während NT-proBNP eine Halbwertszeit von 2 Stunden hat (
30
) und daher stabiler ist (
3
,
12
) und leichter messbar ist (
12
). Die Werte von kardialem und zirkulierendem BNP steigen als Reaktion auf Hypertrophie und/oder ventrikuläre Überlastung signifikant an, was zeigt, dass BNP ein ausgezeichneter Marker für ventrikuläre Dysfunktion ist (
28
).Zur Pathophysiologie der PH: BNP ist bei verschiedenen Formen der PAH erhöht, darunter idiopathische PAH (IPAH) (
32
), PAH in Verbindung mit Bindegewebserkrankungen (CTD-PAH) (
33
), angeborene systemisch-pulmonale Shunts (
34
), PH in Verbindung mit Lungenerkrankungen (
35
), PH und chronisch obstruktiver Lungenerkrankung (COPD) (
28
), chronisch thromboembolische PH (CTEPH) (
29
) und PH in Verbindung mit akuter Lungenembolie (
36
). In mehreren Studien korrelieren BNP-Spiegel eng mit der Funktionsklasse der New York Heart Association (NYHA), dem 6-Minuten-Gehstrecke (6MWD)-Test und hämodynamischen Parametern (
3
,
29
,
32
,
37
). Nagaya et al. es zeigte sich auch, dass BNP-Spiegel positiv mit mPAP und umgekehrt mit dem Herzzeitvolumen korrelieren, wobei eine starke Korrelation mit dem totalen Lungenwiderstand besteht (
38
). NT-proBNP ist auch bei verschiedenen Formen der PAH erhöht, wie etwa bei IPAH (
28
) und bei systemischer Sklerose-assoziierter PAH (SSc-PAH) (
39
). Im letzteren Fall korreliert NT-proBNP mit mPAP, PVR, mittlerem rechtsatrialen Druck (mRAP) und Herzindex (
3
) und kann wirksam als Prädiktor des Überlebens bei PAH dienen (
23
). ANP-Spiegel sind auch bei IPAH (
40
) und CTEPH erhöht. Wie BNP korrelieren ANP-Spiegel positiv mit mPAP und umgekehrt mit dem Herzzeitvolumen (
38
). Allerdings hat ANP bei Menschen eine kurze Halbwertszeit von etwa 2 Minuten (
41
,
42
) - während BNP eine viel längere Halbwertszeit hat (
43
), was BNP zu einem besseren Kandidaten als Biomarker macht (
3
).Somit sind BNP und NT-proBNP sensitive und spezifische Biomarker für die Risikostratifizierung bei PAH (sie sind bislang die einzigen Biomarker, die in den aktuellen Leitlinien enthalten sind) (
23
). Sie sind zudem sensitive Marker für RV-Dysfunktionen und die Wirksamkeit der Behandlung (
23
,
26
,
28
). Zudem besitzt BNP als pulmonaler Vasodilatator und antihypertropher Wirkstoff das therapeutische Potenzial, pulmonalvaskuläre Umbauprozesse zu lindern (
28
). Dabei müssen jedoch immer bestimmte Faktoren berücksichtigt werden, nämlich Geschlecht und Alter des Patienten sowie das Vorliegen einer Linksherzerkrankung, Nierenfunktionsstörungen und Fettleibigkeit (
12
,
26
). In Fällen einer Linksherzerkrankung sollten BNP-Spiegel nicht zur Vorhersage der Diagnose oder Prognose von PAH herangezogen werden (
30
).TroponinKardiale Troponine (cTn) sind eine Gruppe von 3 Proteinen – Troponin C, Troponin I (cTnI) und Troponin T (cTnT) –, deren Hauptfunktion darin besteht, die dünnen Aktinfilamente des Herzmuskels zu regulieren. Der Troponinspiegel hängt eng mit der Schädigung des Myokards zusammen, da das Troponin beim Zerreißen der Kardiomyozytenmembranen ins periphere Blut freigesetzt wird. Daher können sie mittels hochsensibler Tests im Plasma nachgewiesen werden (
12
,
26
). Die Messung von cTnT und cTnI ist von entscheidender Bedeutung für die Diagnose und Prognose von Patienten mit akutem Koronarsyndrom sowie für alle Pathologien, die mit Myokardläsionen einhergehen und erhöhte Werte kardialer Troponine aufweisen, wie z. B. bei Myokarditis (
12
).In der Pathophysiologie der PH: Mehrere Studien haben cTnT mit der schlechten Prognose von PH in Verbindung gebracht. Torbicki et al. zeigten, dass bei Patienten mit PAH und CTEPH die cTnT-Werte erhöht sind, was wahrscheinlich durch die Schädigung des RV-Myokards erklärt werden kann. In dieser Studie hatten jedoch nur 14 % der Patienten (8 von 56 Patienten) erhöhte cTnT-Werte, da der Test zur Messung der cTnT-Werte nur die Erkennung von Werten >0,01 ng/ml ermöglicht. Beim Vergleich von cTnT(+) - und cTnT(-)-Patienten zeigte sich dennoch, dass sie eine ähnliche pulmonale Hämodynamik hatten, aber cTnT(+)-Patienten eine höhere Herzfrequenz, eine geringere gemischtvenöse Sauerstoffsättigung (SvO 2 ), höhere NT-proBNP-Serumwerte und eine geringere Belastungsresistenz (gemessen mit 6MWD) (
44
). Darüber hinaus wurde bei Patienten unter PH-Behandlung festgestellt, dass die cTnT-Spiegel sinken und sogar unter die Nachweisgrenze geraten; im Gegensatz dazu steigen die Werte mit Fortschreiten der Erkrankung an (
44
).Neuere Studien bieten Tests mit höherer Sensitivität gegenüber cTn, aber deutlich niedrigeren Nachweisgrenzen. Heresi et al. zeigten unter Verwendung eines Immunanalysators mit einer Nachweisgrenze von < 0,008 ng/ml, dass bei 25 % der Patienten mit PAH eine Erhöhung des cTnI-Werts festgestellt werden konnte. Im Vergleich zu cTnI(+)-Patienten hatten cTnI(+)-Patienten eine höhere Funktionsklasse, einen größeren rechten Vorhofbereich, eine niedrigere 6MWD und höhere BNP- und C-reaktive Protein- (CRP-) Werte. Zudem war die Überlebensrate von cTnI(+)-Patienten signifikant niedriger als die von cTnI(-)-Patienten (44 % vs. 85 %) (
45
). Filusch et al. untersuchten die cTnT-Werte bei Patienten mit PAH und verglichen den konventionellen Test mit dem hochsensitiven cTnT-Test (hsTnT) mit einer Nachweisgrenze von weniger als 2 pg/ml. Bei 90,9 % der Patienten war cTnT mit dem hsTnT-Test nachweisbar, im Vergleich zu 30,9 % mit dem herkömmlichen Test. Außerdem waren die Messungen des hsTnT-Tests signifikant mit einer systolischen RV-Dysfunktion und einer beeinträchtigten 6MWD verbunden. Darüber hinaus sagte hsTnT eine Funktionsklasse II oder höher der Weltgesundheitsorganisation (WHO) besser voraus als NT-proBNP und sagte den Tod genauso effektiv voraus wie NT-proBNP (
46
). In einer anderen Studie waren mit dem neuen hsTnI-Test cTnI-Werte bei 95 % der Patienten mit PH nachweisbar, einschließlich PAH-Patienten. Höhere cTnI-Werte sind mit höheren BNP-Werten, einer niedrigeren 6MWD, schwerwiegenderen hämodynamischen Anomalien und Anomalien in der kardialen MRT-Bildgebung verbunden (
47
).Obwohl cTn auch mit einigen Markern auf der linken Seite des Herzens in Zusammenhang stehen (
47
), was ein Störfaktor sein kann, wiesen Studien darauf hin, dass sie als Indikatoren für die Schwere der Erkrankung verwendet werden können. Darüber hinaus liefert die Verwendung der neuen hsTn-Tests neue prognostische Informationen, und diese Tests haben das Potenzial, mehr Patienten mit höherem Risiko zu erkennen und so die Risikostratifizierung zu erleichtern. Dennoch gibt es Störfaktoren, die berücksichtigt werden müssen, nämlich das Vorhandensein einer gleichzeitigen Erkrankung der linken Herzhälfte oder eines Nierenversagens (
12
,
26
).Cystatin CCystatin C (CysC) ist ein 13 kDa großes, nicht-glykosyliertes Protein (
48
,
49
), wird von allen untersuchten kernhaltigen Zellen mit konstanter Rate produziert (
50
) und ist ein Mitglied der Cystatin-Superfamilie, die Inhibitoren von Cysteinproteinasen umfasst (
51
). In den letzten Jahren hat CysC aufgrund seiner freien Filtration im Glomerulus, seiner vollständigen Reabsorption und Katabolisierung im proximalen Tubulus und seines Fehlens einer tubulären Sekretion an Bedeutung gewonnen. Man nimmt an, dass die Plasmakonzentration von CysC fast vollständig von der glomerulären Filtrationsrate abhängt, und macht es daher zu einem idealen Marker der Nierenfunktion (
48
,
49
). Außerdem ist es sensitiver als Serumkreatin, da es geringere Verringerungen der glomerulären Filtrationsrate erkennt (
48
,
49
). Mehrere Studien haben gezeigt, dass CysC auch als kardiovaskulärer Risikomarker fungieren kann, da es Linksherzinsuffizienz (HF) und kardiovaskuläre Mortalität im Allgemeinen vorhersagt (
48
,
49
).In der PH-Pathophysiologie: Fenster et al. zeigten, dass Patienten mit PAH abnorm hohe CysC-Serumspiegel haben und dass diese Spiegel mit der RV-Funktion korrelieren (
52
). Diese Studien zeigen, dass der systolische RV-Druck bei PAH-Patienten stark erhöht ist und positiv mit CysC-Serumspiegeln korreliert. Darüber hinaus korrelieren RV-enddiastolisches Volumen, RV-endsystolisches Volumen, Masseindex, Dehnung und Dehnungsrate positiv mit CysC-Spiegeln und die RV-Ejektionsfraktion negativ (
52
). Somit kann CysC ein sensitiver Biomarker zur Beurteilung von PAH sein und hat gegenüber den Standardbiomarkern BNP und NT-proBNT zusätzliche Vorteile, da es unabhängig von Muskelmasse, Alter und Geschlecht ist (
50
).Biomarker der HämatopoeseHomocysteinHomocystein ist ein schwefelhaltiges Zwischenprodukt des normalen Stoffwechsels von Methionin, einer essentiellen Aminosäure, die im Wesentlichen aus tierischem Eiweiß gewonnen wird (
53
). Homocystein muss über einen Weg wiederverwertet werden, der Folsäure und die Vitamine B6 und B12 erfordert . Veränderungen in diesem Stoffwechselweg, wie beispielsweise ein Mangel an Vitamin B6 und B12 , genetische Defekte oder Polymorphismen in den wichtigsten Enzymen dieses Stoffwechselwegs, können zu einem Anstieg des Homocysteinspiegels führen, der schädlich sein kann (
53
). Homocystein ist ein Hemmer der Dimethylarginin-Dimethylaminohydrolase, eines Enzyms, das asymmetrisches Dimethylarginin (ADMA) metabolisiert; ADMA ist ein endogener Hemmer des NO-Synthase(NOS)-Stoffwechselwegs. Daher führt ein Anstieg des Homocysteinspiegels, die sogenannte Hyperhomocysteinämie, zu einer verringerten Bioverfügbarkeit von NO. Man geht davon aus, dass dieser NO-Abnahme darauf zurückzuführen ist, dass Homocystein die Aktivität der Dimethylarginin-Dimethylaminohydrolase hemmt und so die Ansammlung von ADMA fördert und infolgedessen die NO-Produktion durch die endothelialen Zellen verringert (
54
). Darüber hinaus wird angenommen, dass Homocystein am oxidativen Abbau von NO beteiligt ist (
54
,
55
). Da Homocystein im Wesentlichen die endothelialen Zellen, die Blutgerinnung und die Thrombozytenfunktion beeinflusst, erklärt sich, warum die endotheliale Vasodilatatorfunktion bei Personen mit Hyperhomocysteinämie beeinträchtigt und mit einer Thrombozytenfunktionsstörung einhergeht, die die Blutgerinnung erleichtert und somit das Risiko für Herz-Kreislauf-Erkrankungen erhöht und die Entwicklung von Gefäßerkrankungen beschleunigt (
54
,
55
).In der Pathophysiologie der PH zeigen Studien, dass die Gesamthomocysteinwerte im Plasma – einschließlich Homocystein und seiner oxidierten Derivate – gemischtes Homocystein-Cystein-Disulfid und proteingebundenes Homocystein – bei Patienten mit PAH höher sind als bei gesunden Kontrollpersonen (
53
). Darüber hinaus zeigten Sanli et al., dass die Homocysteinwerte bei Patienten mit PAH in Verbindung mit angeborenen Herzfehlern (CHD-PAH) höher sind, ebenso wie die ADMA-Werte, sie korrelierten jedoch nicht mit hämodynamischen Faktoren (
56
). Beide Studien legen also nahe, dass Homocystein ein wichtiger Faktor bei der Entwicklung von PAH sein könnte, was sich durch die Endothelschäden durch Hyperhomocysteinämie gut begründen lässt. Es sollten jedoch weitere Studien durchgeführt werden, da die Stichprobengröße in beiden vorhandenen Studien relativ gering ist (
56
).Verteilungsbreite der roten BlutkörperchenDie Erythrozytenverteilungsbreite (RDW) ist ein Parameter, der die Schwankungen in der Größe der zirkulierenden roten Blutkörperchen widerspiegelt; sie wird routinemäßig bei einem großen Blutbild gemessen und ist von entscheidender Bedeutung für die Differentialdiagnose einer Anämie (
57
–
59
). Mehrere Studien haben gezeigt, dass RDW-Werte bei der Vorhersage bösartiger Tumoren hilfreich sein können (
60
,
61
). Die RDW ist mit verschiedenen pathophysiologischen Mechanismen verbunden, wie Entzündungen, Eisenstoffwechsel, Nierenfunktionsstörungen, Ernährungszustand und oxidativem Stress, die zu einer Verringerung der erythropoetischen Leistung führen (
62
). Erhöhte RDW-Werte stehen in engem Zusammenhang mit einer beeinträchtigten Erythropoese oder dem Abbau von Erythrozyten (
63
). Die RDW hat sich als vielversprechender Prädiktor des klinischen Ausgangs von Nierenerkrankungen, Herz-Kreislauf-Erkrankungen und Lungenerkrankungen (
57
–
59
) wie HF, PH verschiedener Ätiologien (
58
), akutem Myokardinfarkt, ambulant erworbener Pneumonie, Lungenembolie und COPD erwiesen; zudem erwies sie sich als Prädiktor der Mortalität bei Patienten mit COPD und PAH (
64
).In der Pathophysiologie der PH: Ulrich et al. zeigten, dass RDW ein Faktor ist, der mit dem Überleben bei PAH korreliert; und dass bei Patienten mit PAH üblicherweise Eisenmangel auftritt (
65
,
66
). Die RDW-Werte steigen mit sinkenden Eisenwerten, da die verfügbaren Eisenwerte des Körpers nicht dem Eisenbedarf aus der Synthese roter Blutkörperchen entsprechen, was zu unterschiedlichen Größen der roten Blutkörperchen führt (
67
). Rhodes et al. zeigten, dass RDW-Werte mit der WHO-Funktionsklasse und der 6MWD korrelieren (
68
); sie zeigten auch, dass RDW als unabhängiger Prädiktor des Überlebens fungieren kann, auch wenn es in Kombination mit 6MWD, NT-proBNP und anderen klinischen Indizes gemessen wird (
68
).Zusammenfassend lässt sich sagen, dass RDW eng mit der Schwere der Erkrankung zusammenhängt und zur Vorhersage des Überlebens von Patienten mit IPAH verwendet werden kann (
67
,
68
). Daher sollte die Verwendung von RDW als PAH-Biomarker für neue Ansätze mit mehreren Biomarkern zur PAH-Stratifizierung in Betracht gezogen werden, da die RDW-Werte in Kombination mit den NT-proBNP-Werten eine bessere Erkennung von Hochrisikofällen zeigten als die Verwendung von NT-proBNP allein (
68
).Biomarker für endotheliale Dysfunktion und/oder Gefäßumbau und -schädigungEndothelin-1Das ET-System besteht aus drei ET-Isopeptiden (ET-1, ET-2 und ET-3), die Peptidasen mit unterschiedlichen Isoformen aktivieren, und zwei G-Protein-gekoppelten Rezeptoren – dem ET-Typ-A-Rezeptor (ETA) und dem ET-Typ-B-Rezeptor (ETB) (
69
). ETs sind Isopeptide mit 21 Aminosäuren und weisen eine hohe Homologie und Ähnlichkeit zueinander auf. Sie werden hauptsächlich in ECs exprimiert; sie werden jedoch auch in Herzmyozyten, Lungenepithel, glomerulären Nierenzellen, Mesangialzellen, glatten Muskelzellen (SMCs), Leukozyten, Makrophagen (
7
,
70
) und Fibroblasten (
12
) exprimiert. ETs gelten als die wirksamsten endogenen Vasokonstriktoren (
69
), darüber hinaus sind sie aber auch multifunktionelle Peptide mit zytokin- oder hormonähnlicher Aktivität (
71
).ET-1 ist die am stärksten exprimierte Form im Herzkreislaufsystem (
7
) und in den Lungengefäßen (
12
); es bindet an beide Rezeptoren. ETA und ETB sind in verschiedenen Geweben und Zellen verteilt, ihre Expression ist jedoch variabel; ETA wird vorwiegend auf vaskulären glatten Muskelzellen (VSMCs) (
71
) und Myozyten (
7
) exprimiert, während ETB vorwiegend auf ECs (ETB1-Rezeptoren) und VSMCs (ETB2-Rezeptoren) (
7
) lokalisiert ist. Die Aktivierung von ETA vermittelt vor allem Vasokonstriktion sowie Zellproliferation (
69
). Im Gegensatz dazu fördert die Aktivierung von ETB in ECs eine indirekte vasodilatatorische Wirkung, hemmt die Proliferation von Myokard- und Gefäßgewebe und reguliert den Nierenblutdruck. Auf der anderen Seite spielen ETB2-Rezeptoren eine gewisse vasokonstriktorische Rolle, ihre wichtigste Wirkung ist jedoch die ET-1-Clearance (
69
). Die Vasokonstriktion wird durch die Aktivierung von Phospholipase C, einen Anstieg von Inositoltriphosphat und Diacylglycerol und einen anschließenden Anstieg von intrazellulärem Calcium vermittelt, was die Zellkontraktion fördert. Andererseits stimuliert die durch die Aktivierung des endothelialen Rezeptors ETB1 vermittelte Vasodilatation die Freisetzung von NO und Prostacyclinen, was zur Entspannung der Gefäßwand führt (
7
). Die ET-1-Clearance erfolgt durch Internalisierung des Rezeptors, an den ET-1 gebunden hat; die Lunge eliminiert etwa 50 % des zirkulierenden ET-1 (
7
).ET-1 induziert eine intensive und anhaltende Vasokonstriktion der Lungenarterien und -venen, selbst wenn es in niedriger Konzentration vorhanden ist, und kann auch die Proliferation von Lungenfibroblasten stimulieren. Auf kardialer Ebene ist ET-1 an der Steigerung der myokardialen Kontraktilität und der Herzfrequenz beteiligt (positiv inotroper bzw. chronotroper Effekt) und stimuliert darüber hinaus die Produktion von Zytokinen, Wachstumsfaktoren und Matrixproteinen in anderen Geweben (
7
).In der Pathophysiologie der PH weisen die meisten Patienten erhöhte ET-1-Werte auf (
72
). ET-1 wird als Mediator für erhöhten Gefäßtonus und Gefäßumbau angesehen (
73
). Bei PAH kommt es zu einer deutlichen Erhöhung der ET-1-Expression in den Lungengefäßen (
73
), einschließlich der für die Erkrankung charakteristischen plexiformen Läsionen (
7
). Zudem sind die ET-1-Plasmaspiegel erhöht und stehen in enger Korrelation mit RAP und der Sauerstoffsättigung der Lungenarterie (
5
,
74
), PVR und 6MWD (
75
). Eine für PAH charakteristische Endothelschädigung verstärkt die konstriktive Wirkung von ET-1, verursacht eine Dysregulation im ET-System (
7
) und verringert die Fähigkeit des Endothels, Vasodilatatoren freizusetzen (
72
). Diese Dysregulation und Überexpression von ET-1 fördern also einen Anstieg des PVR (
7
), was teilweise auf den Mangel an Vasodilatatoren (
72
) und auf eine abnorme pulmonale Gefäßumgestaltung (
19
) zurückzuführen ist. Hohe ET-1-Konzentrationen sind auch mit einer Entzündungsreaktion und verstärkter Fibrose assoziiert (
1
). Der Anstieg der ET-1-Plasmakonzentrationen bei PAH kann auf eine erhöhte ET-1-Freisetzung oder eine Verringerung der ET-1-Clearance durch das Lungengefäßsystem oder sogar auf eine Kombination beider Faktoren zurückzuführen sein (
1
,
72
).Darüber hinaus ist die Hemmung der ET-Rezeptoren durch ERAs in der Behandlung von PAH effektiv, da sie PAP verringert und die vaskuläre Umgestaltung hemmt. Bosentan zeigte Verbesserungen der hämodynamischen Parameter, der 6MWD und der WHO-Funktionsklasse bei Patienten mit PAH (
76
); Sitaxsentan zeigte Verbesserungen bei 6MWD, WHO-Funktionsklasse, PVR und Herzindex (
77
). Ambrisentan verbessert die körperliche Belastbarkeit, die WHO-Funktionsklasse, die hämodynamischen Parameter und die Sterblichkeit bei PAH-Patienten; zudem ist es mit einem geringen Risiko von Aminotransferaseanomalien verbunden (
78
,
79
). Darüber hinaus zeigte die AMBITION-Studie, dass eine Zweifachkombinationstherapie mit Ambrisentan und Tadalafil, einem PDE-5-Hemmer, das Risiko eines PAH-bedingten Krankenhausaufenthalts im Vergleich zu einer Monotherapie um 63 % senkte (
80
). Macitentan verbessert mPAP, RAP, PVR, Herzindex und die NT-proBNP-Werte (
81
).Daher spielt das ET-1-System eine grundlegende Rolle in der Pathologie der PAH und kann sogar als prognostischer Marker der Krankheit verwendet werden. Die Verwendung von ET-1 als Marker ist jedoch mit einigen Einschränkungen verbunden, die berücksichtigt werden müssen: Da es sich im Wesentlichen über Gefäßstrukturen ausbreitet, spiegeln seine Plasmaspiegel die ET-1-Konzentration im Gewebe nicht genau wider (
12
,
72
). Außerdem müssen demografische Merkmale wie Ethnizität, Geschlecht und Alter immer berücksichtigt werden, da die ET-1-Plasmaspiegel bei Menschen afrikanischer Abstammung, Männern und höherem Alter höher sind und somit potenzielle Störfaktoren darstellen (
82
).AdrenomedullinAdrenomedullin (ADM) ist ein 52 Aminosäuren langes Peptidhormon, das mit einer langanhaltenden pulmonal-vasodilatatorischen Wirkung in Verbindung gebracht wird (
83
,
84
). ADM wurde erstmals 1993 aus menschlichem Phäochromozytom isoliert (
83
,
84
) und später als zirkulierendes Hormon entdeckt (
12
). ADM wird von endothelialen Zellen (ECs) und endothelialen Zellmembranen (VSMCs) produziert und diffundiert zwischen Blut und Interstitium (
83
,
85
). Obwohl es im ganzen Körper Rezeptoren und Bindungsstellen gibt, ist die Rezeptordichte in den Herz-Kreislauf- und Lungengeweben höher, weshalb es hauptsächlich in diesen beiden Systemen wirkt (
83
,
85
). Neben seiner gefäßerweiternden Wirkung hat ADM auch diuretische und natriuretische Wirkungen, hemmt das Renin-Angiotensin-Aldosteron-System und ist an der Angiogenese und der Regulierung von Entzündungen beteiligt (
12
). Aufgrund seiner gefäßerweiternden Wirkung sowie seiner natriuretischen und diuretischen Wirkung ist es verständlich, dass ADM an der Regulierung der Körperflüssigkeit und damit an der Herzhomöostase beteiligt ist (
85
). Außerdem hat es die Fähigkeit, als Regulator des pulmonalen Gefäßtonus und der Gefäßumgestaltung zu wirken (
83
). Frühere Studien zeigten, dass die Plasmaspiegel von ADM bei Patienten mit Bluthochdruck und Herzinsuffizienz erhöht sind.In der Pathophysiologie der PH sind die Plasmaspiegel von ADM bei Patienten mit PAH erhöht und steigen proportional zum Schweregrad der PH an (
83
,
85
). Zudem korrelieren ADM-Spiegel mit klinischen Parametern, einschließlich mRAP-, 6MWD- und NT-proBNP-Spiegeln, mit ESC/ERS- und REVEAL-Risikoscores und können das Gesamtüberleben der Patienten widerspiegeln (
86
). Studien zeigten auch die Bedeutung von ADM als therapeutisches Ziel für PAH – die Gabe von exogenem ADM führte bei Patienten mit PAH zu signifikanten hämodynamischen Verbesserungen (Anstieg des Herzindex und Rückgang des PVR) (
83
); somit wirkt es als krankheitsregulierendes Hormon bei PAH und zusätzlich als alternativer Prognose- und Schweregradmarker (
86
).CopeptinCopeptin ist ein 39 Aminosäuren langes Glykopeptid und ist vor allem als Arginin-Vasopressin (AVP)-assoziiertes Glykopeptid bekannt (
87
). Copeptin wird, wie auch AVP und Neurophysin II, von einem Vorläufer, dem Prä-Pro-Vasopressin, abgeleitet (
87
,
88
). Diese drei Peptide werden stöchiometrisch von der Hypophyse sezerniert, und es ist möglich, Copeptinwerte als Indikator für AVP-Werte zu verwenden (
87
,
88
). AVP wird im Hypothalamus produziert und von der Hypophyse als Reaktion auf hämodynamische und/oder osmotische Reize sezerniert (
87
). AVP bindet an zwei Rezeptortypen: Vasopressin1, das die arterioläre Vasokonstriktion vermittelt (
87
,
88
), und Vasopressin2, das antidiuretisch wirkt (
87
), indem es die Wasserrückresorption durch die Induktion von Aquaporinen in den Sammelrohren der Niere fördert (
88
). AVP hat jedoch eine kurze Plasmahalbwertszeit (
89
), und da es instabil ist, ist das zirkulierende AVP hauptsächlich an Blutplättchen gebunden (
88
) und daher nicht messbar. Copeptin ist ein Prognosemarker für verschiedene kardiovaskuläre Pathologien (
12
,
87
,
88
).In der PH-Pathophysiologie sind die Konzentrationen des zirkulierenden Copeptins bei PAH-Patienten erhöht und korrelieren positiv mit der NYHA-Klasse und negativ mit der 6MWD (
88
). Darüber hinaus wurde festgestellt, dass der Anstieg des Plasmavolumens und niedrige Natriumkonzentrationen im P
- 1 UnIC@RISE, Abteilung für Chirurgie und Physiologie, Medizinische Fakultät der Universität Porto, Porto, Portugal
- 2 Paris-Porto Pulmonary Hypertension Collaborative Laboratory (3PH), UMR_S 999, INSERM, Université Paris-Saclay, Paris, Frankreich
- 3 Université Paris–Saclay, AP-HP, INSERM UMR_S 999, Service de Pneumologie et Soins Intensifs Respiratoires, Hôpital de Bicêtre, Le Kremlin Bicêtre, Frankreich
- 4 Fakultät für Ernährungs- und Lebensmittelwissenschaften, Universität Porto, Porto, Portugal
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.