
- Beiträge: 1755
Sidebar
Pulmonale Hypertonie, Covid
13 Aug 2024 13:20 #2178
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Pulmonale Hypertonie, Covid wurde erstellt von danny
Mögliche therapeutische Ziele für COVID-19 mit pulmonaler Hypertonie: eine bioinformatische und frühe Validierungsstudie
 Abnorme Hochregulierung des Herz-Kreislauf-Biomarkers PLA2G7 durch proinflammatorische Makrophagen bei COVID-19-Patienten
Artikel Offener Zugang24. März 2021
Abnorme Hochregulierung des Herz-Kreislauf-Biomarkers PLA2G7 durch proinflammatorische Makrophagen bei COVID-19-Patienten
Artikel Offener Zugang24. März 2021
 Bioinformatik-basierte Untersuchung des genetischen Einflusses zwischen SARS-CoV-2-Infektionen und Erkrankungen der idiopathischen Lungenfibrose (IPF) sowie der Neuverwendung von Medikamenten
Artikel Offener Zugang22. März 2023
Bioinformatik-basierte Untersuchung des genetischen Einflusses zwischen SARS-CoV-2-Infektionen und Erkrankungen der idiopathischen Lungenfibrose (IPF) sowie der Neuverwendung von Medikamenten
Artikel Offener Zugang22. März 2023
 Transkriptomische Datenuntersuchung von Konsensgenen und molekularen Mechanismen zwischen chronisch obstruktiver Lungenerkrankung und Lungenadenokarzinom
Artikel Offener Zugang02. August 2022EinführungDas Schwere Akute Respiratorische Syndrom Coronavirus 2 (SARS-CoV-2), das für die Coronavirus-Erkrankung (COVID-19) verantwortlich ist, hat sich weltweit verbreitet und wurde am 11. März 2020 zur Pandemie erklärt. Es stellt weltweit eine große Gefahr für die öffentliche Gesundheit dar
1
,
2
,
3.
In den letzten zwei Jahren sind mehrere mutierte Stämme von SARS-CoV-2 aufgetaucht, die das Potenzial des Virus erhöhen, Menschen zu infizieren oder den Impfschutz zu umgehen. COVID-19 wird weiterhin eine weltweite Belastung für das Gesundheitswesen und die Volkswirtschaften darstellen, solange Menschen damit infiziert sind. In der Zeit nach der Pandemie sollten wir uns auch mit den Langzeitfolgen von COVID-19 und seiner Behandlung befassen
4
,
5
,
6
.Langzeitfolgen von COVID-19 können in den Atemwegen und im Herz-Kreislauf-System beobachtet werden, aber das Virus befällt auch das Nervensystem, die Knochen und die endokrinen Drüsen. Wichtige Folgen von COVID-19 sind das akute Atemnotsyndrom (ARDS), venöse Thromboembolien, pulmonale Hypertonie und akute Herzschäden
5
,
6
.Es wurde berichtet, dass pulmonale Hypertonie den Krankheitsverlauf bei 13,4 % bzw. 21 % der Patienten mit einer Infektion mit dem neuen Coronavirus kompliziert. Pulmonale Hypertonie ist eine ernste Komplikation einer Infektion mit dem neuen Coronavirus und erhöht die Wahrscheinlichkeit, dass eine Behandlung auf der Intensivstation, künstliche Beatmung, extrakorporale Membranoxygenierung (ECMO) oder sogar der Tod erforderlich werden. Daher könnte das frühzeitige Erkennen von hohem pulmonalarteriellem Druck bei SARS-CoV-2-Patienten die Langzeitprognose der Patienten verbessern und die Hospitalisierungsrate sowie die Sterberate aufgrund derartiger Komplikationen minimieren
7
,
8
,
9
,
10.
Wie frühere Studien gezeigt haben, können die Prozesse der immunologischen Dysfunktion, endothelialen Dysfunktion, Gefäßleckage und thrombotischen Mikroangiopathie, die mit denen vergleichbar sind, die pulmonale Gefäßerkrankungen verursachen, für die Auswirkungen von SARS-CoV-2 auf die pulmonale Hämodynamik verantwortlich sein. Andererseits sind Berichte über die Tiefe und Spezifität des Studienmechanismus selten
7
,
8
,
9
,
10
,
11.
Ziel dieser Untersuchung ist es, mehr über den Zusammenhang zwischen COVID-19 und pulmonaler Hypertonie zu erfahren, indem die pathogenen Moleküle, pathologischen Prozesse und potenziellen therapeutischen Ziele untersucht werden.Hier haben wir eine funktionelle Anreicherungsanalyse der GEO-Datenbank durchgeführt, um häufige differentiell exprimierte Gene (C-DEGs) in den COVID-19- und PH-Datensätzen zu identifizieren. Die Validierungswarteschlange bestätigte die Ergebnisse eines Screenings der Schlüsselgene mithilfe von drei maschinellen Algorithmen: LASSO, RF und SVM-RFE-basiert. Die Gensatzanreicherungsanalyse (GSEA) wurde verwendet, um die Rolle priorisierter Kerngene zu untersuchen. Als nächstes haben wir die regulatorischen Netzwerke einschließlich dieser DEGs kartiert, einschließlich TF-Genverbindungen und TF-microRNA-Koregulierung. Arzneimittel-Protein-Interaktionsnetzwerke, molekulare Dockingsimulationen und molekulare Dynamiksimulationen wurden eingesetzt, um nach möglichen therapeutischen Medikamenten zu suchen. Unsere Erkenntnisse werden voraussichtlich einen neuartigen Ansatz zur Aufklärung der genetischen Verbindung zwischen den oben genannten Erkrankungen bieten. Abbildung
1
zeigt unser Forschungsprotokoll.Abbildung 1
Transkriptomische Datenuntersuchung von Konsensgenen und molekularen Mechanismen zwischen chronisch obstruktiver Lungenerkrankung und Lungenadenokarzinom
Artikel Offener Zugang02. August 2022EinführungDas Schwere Akute Respiratorische Syndrom Coronavirus 2 (SARS-CoV-2), das für die Coronavirus-Erkrankung (COVID-19) verantwortlich ist, hat sich weltweit verbreitet und wurde am 11. März 2020 zur Pandemie erklärt. Es stellt weltweit eine große Gefahr für die öffentliche Gesundheit dar
1
,
2
,
3.
In den letzten zwei Jahren sind mehrere mutierte Stämme von SARS-CoV-2 aufgetaucht, die das Potenzial des Virus erhöhen, Menschen zu infizieren oder den Impfschutz zu umgehen. COVID-19 wird weiterhin eine weltweite Belastung für das Gesundheitswesen und die Volkswirtschaften darstellen, solange Menschen damit infiziert sind. In der Zeit nach der Pandemie sollten wir uns auch mit den Langzeitfolgen von COVID-19 und seiner Behandlung befassen
4
,
5
,
6
.Langzeitfolgen von COVID-19 können in den Atemwegen und im Herz-Kreislauf-System beobachtet werden, aber das Virus befällt auch das Nervensystem, die Knochen und die endokrinen Drüsen. Wichtige Folgen von COVID-19 sind das akute Atemnotsyndrom (ARDS), venöse Thromboembolien, pulmonale Hypertonie und akute Herzschäden
5
,
6
.Es wurde berichtet, dass pulmonale Hypertonie den Krankheitsverlauf bei 13,4 % bzw. 21 % der Patienten mit einer Infektion mit dem neuen Coronavirus kompliziert. Pulmonale Hypertonie ist eine ernste Komplikation einer Infektion mit dem neuen Coronavirus und erhöht die Wahrscheinlichkeit, dass eine Behandlung auf der Intensivstation, künstliche Beatmung, extrakorporale Membranoxygenierung (ECMO) oder sogar der Tod erforderlich werden. Daher könnte das frühzeitige Erkennen von hohem pulmonalarteriellem Druck bei SARS-CoV-2-Patienten die Langzeitprognose der Patienten verbessern und die Hospitalisierungsrate sowie die Sterberate aufgrund derartiger Komplikationen minimieren
7
,
8
,
9
,
10.
Wie frühere Studien gezeigt haben, können die Prozesse der immunologischen Dysfunktion, endothelialen Dysfunktion, Gefäßleckage und thrombotischen Mikroangiopathie, die mit denen vergleichbar sind, die pulmonale Gefäßerkrankungen verursachen, für die Auswirkungen von SARS-CoV-2 auf die pulmonale Hämodynamik verantwortlich sein. Andererseits sind Berichte über die Tiefe und Spezifität des Studienmechanismus selten
7
,
8
,
9
,
10
,
11.
Ziel dieser Untersuchung ist es, mehr über den Zusammenhang zwischen COVID-19 und pulmonaler Hypertonie zu erfahren, indem die pathogenen Moleküle, pathologischen Prozesse und potenziellen therapeutischen Ziele untersucht werden.Hier haben wir eine funktionelle Anreicherungsanalyse der GEO-Datenbank durchgeführt, um häufige differentiell exprimierte Gene (C-DEGs) in den COVID-19- und PH-Datensätzen zu identifizieren. Die Validierungswarteschlange bestätigte die Ergebnisse eines Screenings der Schlüsselgene mithilfe von drei maschinellen Algorithmen: LASSO, RF und SVM-RFE-basiert. Die Gensatzanreicherungsanalyse (GSEA) wurde verwendet, um die Rolle priorisierter Kerngene zu untersuchen. Als nächstes haben wir die regulatorischen Netzwerke einschließlich dieser DEGs kartiert, einschließlich TF-Genverbindungen und TF-microRNA-Koregulierung. Arzneimittel-Protein-Interaktionsnetzwerke, molekulare Dockingsimulationen und molekulare Dynamiksimulationen wurden eingesetzt, um nach möglichen therapeutischen Medikamenten zu suchen. Unsere Erkenntnisse werden voraussichtlich einen neuartigen Ansatz zur Aufklärung der genetischen Verbindung zwischen den oben genannten Erkrankungen bieten. Abbildung
1
zeigt unser Forschungsprotokoll.Abbildung 1
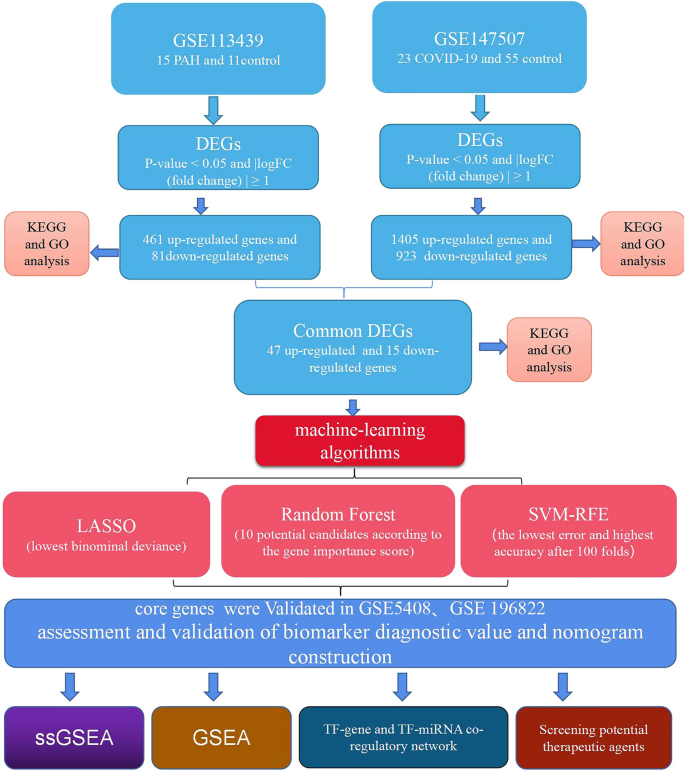 Flussdiagramm des Forschungsdesigns.
ErgebnisseCOVID- und PH-basiertes DEGs-ScreeningIm GSE147507-Datensatz gab es 1405 unterschiedlich exprimierte Gene (Abb.
2
A) und 923 nicht unterschiedlich exprimierte Gene (Abb.
2
Flussdiagramm des Forschungsdesigns.
ErgebnisseCOVID- und PH-basiertes DEGs-ScreeningIm GSE147507-Datensatz gab es 1405 unterschiedlich exprimierte Gene (Abb.
2
A) und 923 nicht unterschiedlich exprimierte Gene (Abb.
2
") zwischen COVID-19-Patienten und Kontrollen. Wie in Abb. 2B gezeigt werden kann, konnten im GSE113439-Datensatz 461 stark exprimierte und 81 kaum exprimierte Gene beobachtet werden, die PH-Patienten von gesunden Kontrollen unterscheiden. Mithilfe des KEGG fanden wir heraus, dass der PH-Datensatz angereichert war mit dem Prozess der Regulierung des Aktin-Zytoskeletts (Padj = 0,031), der Ribosomenbiogenese in Eukaryoten (Padj = 0,0073), des RNA-Abbaus (Padj = 0,0072), des nukleozytoplasmatischen Transports (Padj = 0,022), der Kontraktion der Gefäßglattmuskulatur (Padj = 0,038) und der Motorproteine (Padj = 0,022). Es lag eine hypertrophe Kardiomyopathie (Padj = 0,014), eine Spondyloarthritis (Padj = 0,017) und eine rechtsventrikuläre Kardiomyopathie aufgrund von Arrhythmie (Padj = 0,022) vor (Abb.
2
C). Der COVID-19-Datensatz hingegen wurde angereichert mit einer Infektion mit Herpes-simplex-Virus 1 (Padj = 0,000053), einer Interaktion zwischen Zytokinen und ihren Rezeptoren (Padj = 0,00000002), einer Interaktion zwischen neuroaktiven Liganden und Rezeptoren (Padj = 0,00086), einem PI3K-Akt-Signalweg (Padj = 0,016), Lipid- und atherosklerotischen Erkrankungen (Padj = 0,000044), Tuberkulose (Padj = 0,00000083), einem Chemokin-Signalweg (Padj = 0,0000051), einem NOD-ähnlichen Rezeptor-Signalweg (Padj = 0,000013) und COVID-19 (Padj = 0,025) (Abb.
2
D). Darüber hinaus wurden bei der Analyse von Genen unter Verwendung von GO-Begriffen folgende Anreicherungen von DEGs im COVID-Datensatz als Reaktion auf eine Entzündungsreaktion (Padj = 7,20 × 10–26 ) , eine Immunreaktion (Padj = 8,40 × 10–16 ) , eine Chemotaxis von Neutrophilen (Padj = 4,00 × 10–14 ) , eine Zell-Zell-Signalisierung (Padj = 8,30 × 10–12 ) und eine zelluläre Reaktion auf Lipopolysaccharide (Padj = 1,30 × 10–11 ) beobachtet (Abb.
2
E). Die Förderung von RNA-Polymerase II (Padj = 2,30 × 10–03 ) , Zellteilung (Padj = 1,30 × 10–05 ) , Proteinphosphorylierung (Padj = 8,50 × 10–03 ) , Zelladhäsion (Padj = 0,015), zellulärer Reaktion auf DNA-Schäden (Padj = 3,30 × 10–06 ) , DNA-Reparatur (Padj = 4,40 × 10–06 ) , Entzündungsreaktion (Padj = 0,005) und negativer Regulation des apoptotischen Prozesses (Padj = 0,038) wurden im PH-Datensatz angereichert, wie in Abb.
2
F gezeigt.Abbildung 2
zwischen COVID-19-Patienten und Kontrollen. Wie in Abb. 2B gezeigt werden kann, konnten im GSE113439-Datensatz 461 stark exprimierte und 81 kaum exprimierte Gene beobachtet werden, die PH-Patienten von gesunden Kontrollen unterscheiden. Mithilfe des KEGG fanden wir heraus, dass der PH-Datensatz angereichert war mit dem Prozess der Regulierung des Aktin-Zytoskeletts (Padj = 0,031), der Ribosomenbiogenese in Eukaryoten (Padj = 0,0073), des RNA-Abbaus (Padj = 0,0072), des nukleozytoplasmatischen Transports (Padj = 0,022), der Kontraktion der Gefäßglattmuskulatur (Padj = 0,038) und der Motorproteine (Padj = 0,022). Es lag eine hypertrophe Kardiomyopathie (Padj = 0,014), eine Spondyloarthritis (Padj = 0,017) und eine rechtsventrikuläre Kardiomyopathie aufgrund von Arrhythmie (Padj = 0,022) vor (Abb.
2
C). Der COVID-19-Datensatz hingegen wurde angereichert mit einer Infektion mit Herpes-simplex-Virus 1 (Padj = 0,000053), einer Interaktion zwischen Zytokinen und ihren Rezeptoren (Padj = 0,00000002), einer Interaktion zwischen neuroaktiven Liganden und Rezeptoren (Padj = 0,00086), einem PI3K-Akt-Signalweg (Padj = 0,016), Lipid- und atherosklerotischen Erkrankungen (Padj = 0,000044), Tuberkulose (Padj = 0,00000083), einem Chemokin-Signalweg (Padj = 0,0000051), einem NOD-ähnlichen Rezeptor-Signalweg (Padj = 0,000013) und COVID-19 (Padj = 0,025) (Abb.
2
D). Darüber hinaus wurden bei der Analyse von Genen unter Verwendung von GO-Begriffen folgende Anreicherungen von DEGs im COVID-Datensatz als Reaktion auf eine Entzündungsreaktion (Padj = 7,20 × 10–26 ) , eine Immunreaktion (Padj = 8,40 × 10–16 ) , eine Chemotaxis von Neutrophilen (Padj = 4,00 × 10–14 ) , eine Zell-Zell-Signalisierung (Padj = 8,30 × 10–12 ) und eine zelluläre Reaktion auf Lipopolysaccharide (Padj = 1,30 × 10–11 ) beobachtet (Abb.
2
E). Die Förderung von RNA-Polymerase II (Padj = 2,30 × 10–03 ) , Zellteilung (Padj = 1,30 × 10–05 ) , Proteinphosphorylierung (Padj = 8,50 × 10–03 ) , Zelladhäsion (Padj = 0,015), zellulärer Reaktion auf DNA-Schäden (Padj = 3,30 × 10–06 ) , DNA-Reparatur (Padj = 4,40 × 10–06 ) , Entzündungsreaktion (Padj = 0,005) und negativer Regulation des apoptotischen Prozesses (Padj = 0,038) wurden im PH-Datensatz angereichert, wie in Abb.
2
F gezeigt.Abbildung 2
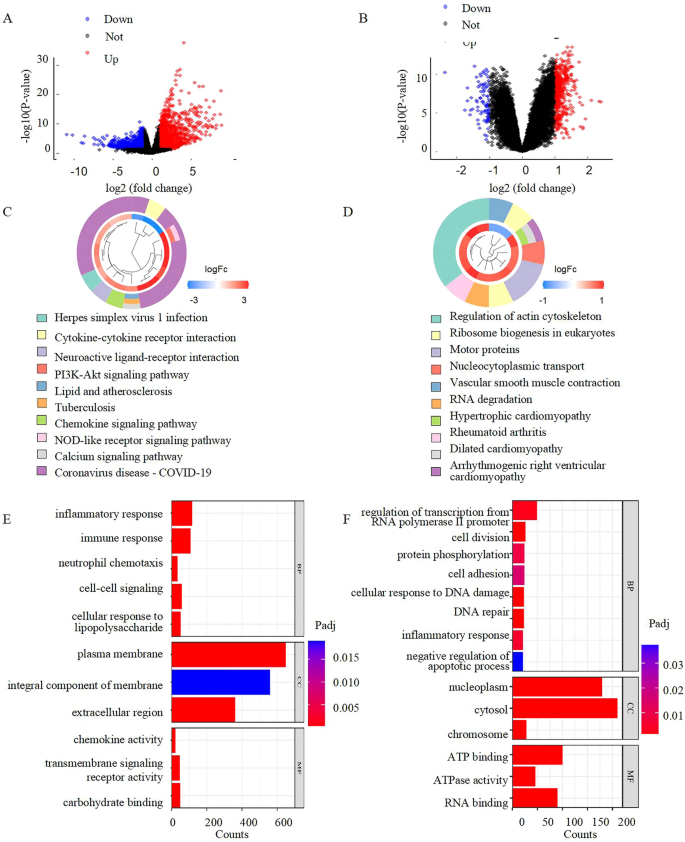 Screening-DEGs für COVID-19 bzw. PH. ( A ) Vulkandiagramm zwischen COVID-19-Patienten und gesunden Kontrollpersonen. 1405 Up-Gene (rot) und 923 Down-Gene (blau). ( B ) Vulkandiagramm zwischen PH-Patienten und gesunden Kontrollpersonen. 461 Up-Gene (rot) und 81 Down-Gene (blau). ( C , D ) Die KEGG-Analyse der COVID-19-DEGs, PH-DEGs. ( E , F ) Die GO-Analyse der COVID-19-DEGs, PH-DEGs. COVID-19- Coronavirus-Krankheit, PH- Lungenhypertonie, DEG- differenziell exprimierte Gene, KEGG Kyoto Encyclopedia of Genes and Genomes, GO Gene Ontology.
C-DEGs-ScreeningMithilfe eines Venn-Diagramms zum Vergleich der DEGs in den Datensätzen GSE147507 und GSE113439 fanden wir 62 C-DEGs (Ergänzende Tabelle
S1
), von denen 47 signifikant hochreguliert (Abb.
3
A) und 15 herunterreguliert (Abb.
3
waren.Abbildung 3
Screening-DEGs für COVID-19 bzw. PH. ( A ) Vulkandiagramm zwischen COVID-19-Patienten und gesunden Kontrollpersonen. 1405 Up-Gene (rot) und 923 Down-Gene (blau). ( B ) Vulkandiagramm zwischen PH-Patienten und gesunden Kontrollpersonen. 461 Up-Gene (rot) und 81 Down-Gene (blau). ( C , D ) Die KEGG-Analyse der COVID-19-DEGs, PH-DEGs. ( E , F ) Die GO-Analyse der COVID-19-DEGs, PH-DEGs. COVID-19- Coronavirus-Krankheit, PH- Lungenhypertonie, DEG- differenziell exprimierte Gene, KEGG Kyoto Encyclopedia of Genes and Genomes, GO Gene Ontology.
C-DEGs-ScreeningMithilfe eines Venn-Diagramms zum Vergleich der DEGs in den Datensätzen GSE147507 und GSE113439 fanden wir 62 C-DEGs (Ergänzende Tabelle
S1
), von denen 47 signifikant hochreguliert (Abb.
3
A) und 15 herunterreguliert (Abb.
3
waren.Abbildung 3
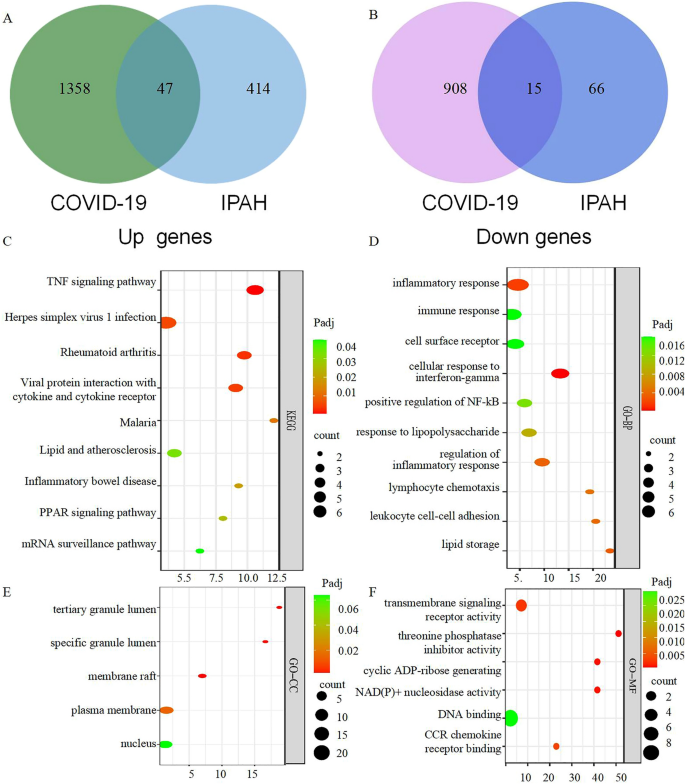 Identifizierung und funktionelle Anreicherungsanalyse häufiger DEGs. ( A ) Venn-Diagramm, das 47 hochregulierte häufige DEGs bei COVID-19 und PH zeigt. ( B ) Venn-Diagramm, das 17 herunterregulierte häufige DEGs bei COVID-19 und PH zeigt. ( C ) An häufigen DEGs wurde eine KEGG-Signalweganalyse durchgeführt. ( D ) GO-BP-Begriffe häufiger Gene. ( E ) GO-CC-Begriffe häufiger Gene. ( F ) GO-MF-Begriffe häufiger Gene. DEG: Differenziell exprimierte Gene. COVID: Coronavirus-Erkrankung. PH: pulmonale Hypertonie. KEGG: Kyoto Encyclopedia of Genes and Genomes. GO-BP-, GO-CC- und GO-MF- Begriffe der Genontologie in biologischen Prozessen, Zellkomponenten und molekularen Funktionen.
Auswertung der C-DEG-FunktionsprofileDie C-DEGs zeigten eine Anreicherung im TNF-Signalweg (Padj = 5,00 × 10–4 ) , bei einer Infektion mit Herpes-simplex-Virus 1 (Padj = 0,0057), bei rheumatoider Arthritis (Padj = 0,4 × 10–3), bei der Interaktion viraler Proteine mit Zytokinen und Zytokinrezeptoren (Padj = 0,0042), bei Malaria (Padj = 0,012), bei Lipiden und Arteriosklerose (Padj = 0,033), bei entzündlichen Darmerkrankungen (Padj = 0,019) und im PPAR-Signalweg (Padj = 0,025) (Abb.
3C
, Zusatztabelle
S2
).Die Untersuchung der Anreicherung des biologischen Prozesses (BP) von GO (Abb.
3
D, Zusatztabelle
S3
) ergab, dass C-DEGs für die folgenden Prozesse angereichert waren: Entzündungsreaktion (Padj = 0,0019), Immunreaktion (Padj = 0,017), Zelloberflächenrezeptor (Padj = 0,017), zelluläre Reaktion auf Interferon-gamma (Padj = 0,00023), positive Regulierung von NF-kB (Padj = 0,014) und Reaktion auf Lipopolysaccharide (Padj = 0,0094). Anreicherungsanalysen für Membranfloß (Padj = 0,00073), tertiäres Granulalumen (Padj = 0,00055) und spezifisches Granulalumen (Padj = 0,00078), transmembranäre Signalrezeptoraktivität (Padj = 0,0025), Threoninphosphatase-Inhibitoraktivität (Padj = 0,00068) und zyklische ADP-Ribose-Erzeugung (Padj = 0,001) wurden an C-DEGs durchgeführt (Abb.
3
E,F). Diese Ergebnisse legen nahe, dass Immunentzündungen eine entscheidende Rolle bei der Ätiologie und dem Fortschreiten von COVID-19 in Verbindung mit PH spielen.Auswahl von Kandidaten für diagnostische Biomarker mittels maschinellem LernenUnter diesen 62DEGs (Ergänzende Tabelle
S4
) identifizierte die LASSO-Regressionsanalyse 20 Gene mit der geringsten binominalen Abweichung (Abb.
4
A,. Der Random-Forest-Ansatz wurde verwendet, um 10 Kandidaten zu identifizieren, nachdem die DEGs nach Gensignifikanzbewertung eingestuft wurden (Abb.
4
C,D). Für die PH-Progression bei COVID19 identifizierte die SVM-RFE-Methode 8 Gene mit dem geringsten Fehler und der besten Genauigkeit nach 100-facher Verwendung (Abb.
4
E,F). Schließlich wurde ein Venn-Diagramm verwendet, um zu zeigen, wie sich SELE und CCL20 (Abb.
4
G) als DEGs erwiesen, wenn beide Methoden zusammen verwendet wurden.Abbildung 4
Identifizierung und funktionelle Anreicherungsanalyse häufiger DEGs. ( A ) Venn-Diagramm, das 47 hochregulierte häufige DEGs bei COVID-19 und PH zeigt. ( B ) Venn-Diagramm, das 17 herunterregulierte häufige DEGs bei COVID-19 und PH zeigt. ( C ) An häufigen DEGs wurde eine KEGG-Signalweganalyse durchgeführt. ( D ) GO-BP-Begriffe häufiger Gene. ( E ) GO-CC-Begriffe häufiger Gene. ( F ) GO-MF-Begriffe häufiger Gene. DEG: Differenziell exprimierte Gene. COVID: Coronavirus-Erkrankung. PH: pulmonale Hypertonie. KEGG: Kyoto Encyclopedia of Genes and Genomes. GO-BP-, GO-CC- und GO-MF- Begriffe der Genontologie in biologischen Prozessen, Zellkomponenten und molekularen Funktionen.
Auswertung der C-DEG-FunktionsprofileDie C-DEGs zeigten eine Anreicherung im TNF-Signalweg (Padj = 5,00 × 10–4 ) , bei einer Infektion mit Herpes-simplex-Virus 1 (Padj = 0,0057), bei rheumatoider Arthritis (Padj = 0,4 × 10–3), bei der Interaktion viraler Proteine mit Zytokinen und Zytokinrezeptoren (Padj = 0,0042), bei Malaria (Padj = 0,012), bei Lipiden und Arteriosklerose (Padj = 0,033), bei entzündlichen Darmerkrankungen (Padj = 0,019) und im PPAR-Signalweg (Padj = 0,025) (Abb.
3C
, Zusatztabelle
S2
).Die Untersuchung der Anreicherung des biologischen Prozesses (BP) von GO (Abb.
3
D, Zusatztabelle
S3
) ergab, dass C-DEGs für die folgenden Prozesse angereichert waren: Entzündungsreaktion (Padj = 0,0019), Immunreaktion (Padj = 0,017), Zelloberflächenrezeptor (Padj = 0,017), zelluläre Reaktion auf Interferon-gamma (Padj = 0,00023), positive Regulierung von NF-kB (Padj = 0,014) und Reaktion auf Lipopolysaccharide (Padj = 0,0094). Anreicherungsanalysen für Membranfloß (Padj = 0,00073), tertiäres Granulalumen (Padj = 0,00055) und spezifisches Granulalumen (Padj = 0,00078), transmembranäre Signalrezeptoraktivität (Padj = 0,0025), Threoninphosphatase-Inhibitoraktivität (Padj = 0,00068) und zyklische ADP-Ribose-Erzeugung (Padj = 0,001) wurden an C-DEGs durchgeführt (Abb.
3
E,F). Diese Ergebnisse legen nahe, dass Immunentzündungen eine entscheidende Rolle bei der Ätiologie und dem Fortschreiten von COVID-19 in Verbindung mit PH spielen.Auswahl von Kandidaten für diagnostische Biomarker mittels maschinellem LernenUnter diesen 62DEGs (Ergänzende Tabelle
S4
) identifizierte die LASSO-Regressionsanalyse 20 Gene mit der geringsten binominalen Abweichung (Abb.
4
A,. Der Random-Forest-Ansatz wurde verwendet, um 10 Kandidaten zu identifizieren, nachdem die DEGs nach Gensignifikanzbewertung eingestuft wurden (Abb.
4
C,D). Für die PH-Progression bei COVID19 identifizierte die SVM-RFE-Methode 8 Gene mit dem geringsten Fehler und der besten Genauigkeit nach 100-facher Verwendung (Abb.
4
E,F). Schließlich wurde ein Venn-Diagramm verwendet, um zu zeigen, wie sich SELE und CCL20 (Abb.
4
G) als DEGs erwiesen, wenn beide Methoden zusammen verwendet wurden.Abbildung 4
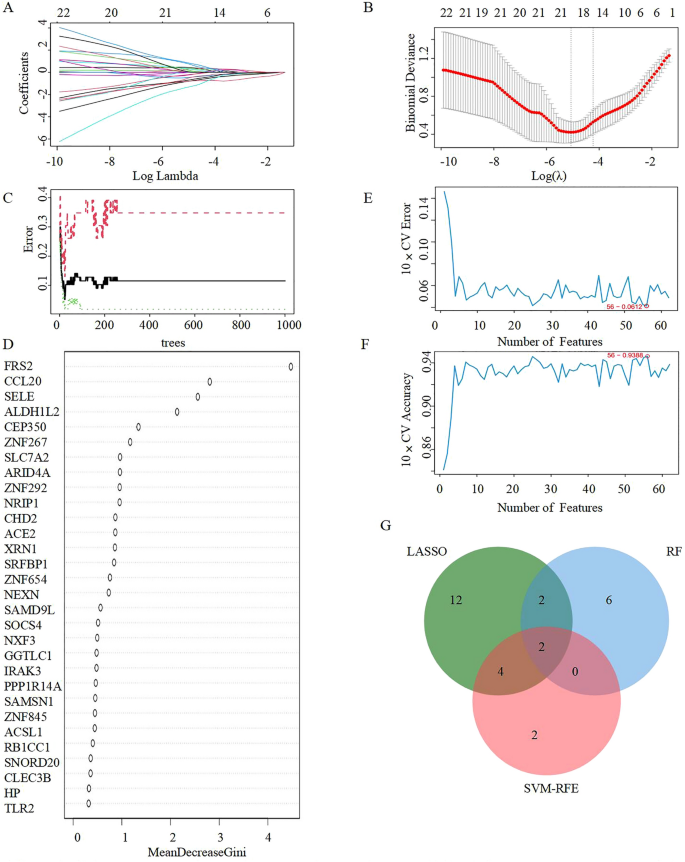 Auswahl von Kandidaten für diagnostische Biomarker für den Verlauf von COVID-19 mit Ansätzen des maschinellen Lernens. ( A , B ) Die LASSO-Regressionsanalyse wurde angewendet, um diagnostische Biomarker zu screenen. ( C ) Der diagnostische Fehler in Bezug auf die Kontroll-, COVID-19- und Gesamtgruppen wurde anhand des Random Forest visualisiert. ( D ) Die Spalte zeigt 30 DEGs, bewertet nach dem Wichtigkeitswert, der aus dem Random Forest berechnet wurde. ( E , F ) Die Anzahl der DEGs mit dem niedrigsten Fehler und der höchsten Genauigkeit nach 100-facher Faltung wurde mithilfe des SVM-RFE-Algorithmus als die am besten geeigneten Kandidaten betrachtet. ( G ) Die Schnittmenge aus 3 Algorithmen des maschinellen Lernens wurde mit einem Venn-Diagramm-Tool ermittelt. LASSO- Operator für kleinste absolute Schrumpfung und Auswahl, SVM-RFE- Support-Vektor-Maschine mit rekursiver Merkmalseliminierung, DEGs – unterschiedlich exprimierte Gene.
Bewertung und Validierung des diagnostischen Werts von Biomarkern und NomogrammerstellungAls Validierungsdatensatz wurden GSE53408 und GSE196822 verwendet. Es gab einen statistisch signifikanten Unterschied in der Expression von SELE und CCL20 zwischen der pulmonalen Hypertonie- und der Kontrollgruppe sowie zwischen der COVID-19- und der Kontrollgruppe (Abb.
5
A,. Nach mehreren Auswahliterationen wurden SELE und CCL20 ausgewählt, um ein Nomogramm zu bilden (Abb.
5
C). Der Expressionsgrad jedes Gens wurde im Nomogramm mit einem numerischen Wert versehen. Schließlich wurde der Gesamtwert angewendet, um die Inzidenz des PH-Fortschritts bei COVID-19-Patienten vorherzusagen (Abb.
5 D). Das Nomogramm zeigte mit einem AUC von 0,826 (95 % KI 0,637–1.000; Abb.
5
D) außergewöhnlich gute Ergebnisse bei der Vorhersage der PH-Entwicklung bei COVID19. Darüber hinaus wurden auch ein Präzisions-Recall (PR) und eine Entscheidungskurvenanalyse (DCA) für das Nomogramm durchgeführt. Diese zeigten, dass das Nomogrammmodell für die Diagnose von COVID-19 mit PH von Nutzen sein kann (Abb.
5
E,F).Abbildung 5
Auswahl von Kandidaten für diagnostische Biomarker für den Verlauf von COVID-19 mit Ansätzen des maschinellen Lernens. ( A , B ) Die LASSO-Regressionsanalyse wurde angewendet, um diagnostische Biomarker zu screenen. ( C ) Der diagnostische Fehler in Bezug auf die Kontroll-, COVID-19- und Gesamtgruppen wurde anhand des Random Forest visualisiert. ( D ) Die Spalte zeigt 30 DEGs, bewertet nach dem Wichtigkeitswert, der aus dem Random Forest berechnet wurde. ( E , F ) Die Anzahl der DEGs mit dem niedrigsten Fehler und der höchsten Genauigkeit nach 100-facher Faltung wurde mithilfe des SVM-RFE-Algorithmus als die am besten geeigneten Kandidaten betrachtet. ( G ) Die Schnittmenge aus 3 Algorithmen des maschinellen Lernens wurde mit einem Venn-Diagramm-Tool ermittelt. LASSO- Operator für kleinste absolute Schrumpfung und Auswahl, SVM-RFE- Support-Vektor-Maschine mit rekursiver Merkmalseliminierung, DEGs – unterschiedlich exprimierte Gene.
Bewertung und Validierung des diagnostischen Werts von Biomarkern und NomogrammerstellungAls Validierungsdatensatz wurden GSE53408 und GSE196822 verwendet. Es gab einen statistisch signifikanten Unterschied in der Expression von SELE und CCL20 zwischen der pulmonalen Hypertonie- und der Kontrollgruppe sowie zwischen der COVID-19- und der Kontrollgruppe (Abb.
5
A,. Nach mehreren Auswahliterationen wurden SELE und CCL20 ausgewählt, um ein Nomogramm zu bilden (Abb.
5
C). Der Expressionsgrad jedes Gens wurde im Nomogramm mit einem numerischen Wert versehen. Schließlich wurde der Gesamtwert angewendet, um die Inzidenz des PH-Fortschritts bei COVID-19-Patienten vorherzusagen (Abb.
5 D). Das Nomogramm zeigte mit einem AUC von 0,826 (95 % KI 0,637–1.000; Abb.
5
D) außergewöhnlich gute Ergebnisse bei der Vorhersage der PH-Entwicklung bei COVID19. Darüber hinaus wurden auch ein Präzisions-Recall (PR) und eine Entscheidungskurvenanalyse (DCA) für das Nomogramm durchgeführt. Diese zeigten, dass das Nomogrammmodell für die Diagnose von COVID-19 mit PH von Nutzen sein kann (Abb.
5
E,F).Abbildung 5
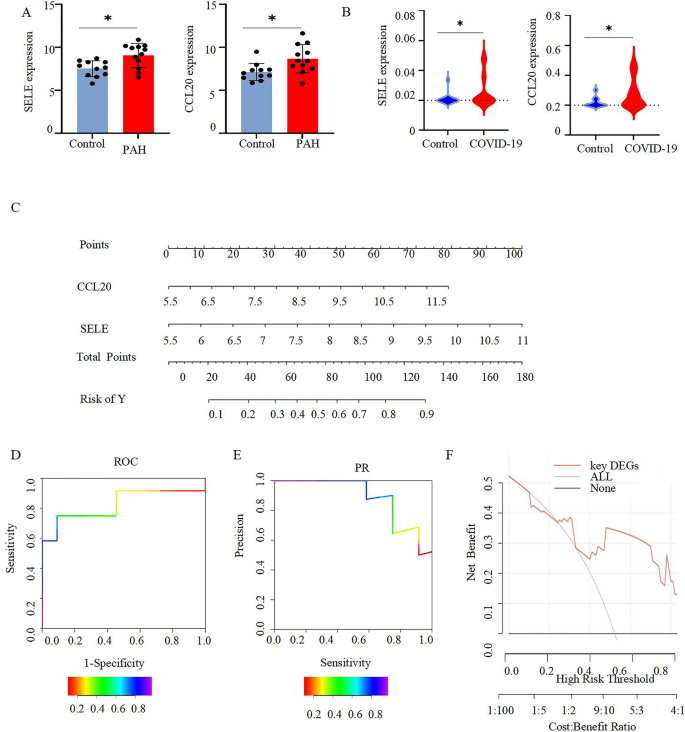 Erstellung eines Nomogramms und Validierung der diagnostischen Leistung. ( A ) SELE, CCL20 wurden in GSE53408 validiert.*P < 0,05. ( B ) SELE, CCL20 wurden in GSE196822 validiert.*P < 0,05. ( C ). Das Nomogramm wurde auf Grundlage der 2 ausgewählten Kandidaten-Biomarker erstellt. Jeder DEG entspricht einem Score. ( D ) Die ROC-Kurve des Nomogramms. ( E ) PR-Analysekurve ( F ) Entscheidungskurvenanalyse (DCA).
GSEA von Hub-GenenWir haben GSEA verwendet, um die wahrscheinlichen physiologischen Funktionen der beiden Hub-Gene zwischen COVID-19 und PH zu bestimmen. Wir fanden heraus, dass erhöhte SELE- und CCL20-Expressionen in GSE113439 (Abb.
6
A, und GSE147507 (Abb.
6
C,D) stark mit aktivierten Immunreaktionen wie der adaptiven Immunreaktion um Blutgefäße herum verbunden waren. Der Anstieg des proinflammatorischen Faktors erhöhte die Expression von Zytokinen und Chemokinen, was zu einer übermäßigen Kontraktion und Proliferation von Blutgefäßzellen beitrug.Abbildung 6
Erstellung eines Nomogramms und Validierung der diagnostischen Leistung. ( A ) SELE, CCL20 wurden in GSE53408 validiert.*P < 0,05. ( B ) SELE, CCL20 wurden in GSE196822 validiert.*P < 0,05. ( C ). Das Nomogramm wurde auf Grundlage der 2 ausgewählten Kandidaten-Biomarker erstellt. Jeder DEG entspricht einem Score. ( D ) Die ROC-Kurve des Nomogramms. ( E ) PR-Analysekurve ( F ) Entscheidungskurvenanalyse (DCA).
GSEA von Hub-GenenWir haben GSEA verwendet, um die wahrscheinlichen physiologischen Funktionen der beiden Hub-Gene zwischen COVID-19 und PH zu bestimmen. Wir fanden heraus, dass erhöhte SELE- und CCL20-Expressionen in GSE113439 (Abb.
6
A, und GSE147507 (Abb.
6
C,D) stark mit aktivierten Immunreaktionen wie der adaptiven Immunreaktion um Blutgefäße herum verbunden waren. Der Anstieg des proinflammatorischen Faktors erhöhte die Expression von Zytokinen und Chemokinen, was zu einer übermäßigen Kontraktion und Proliferation von Blutgefäßzellen beitrug.Abbildung 6
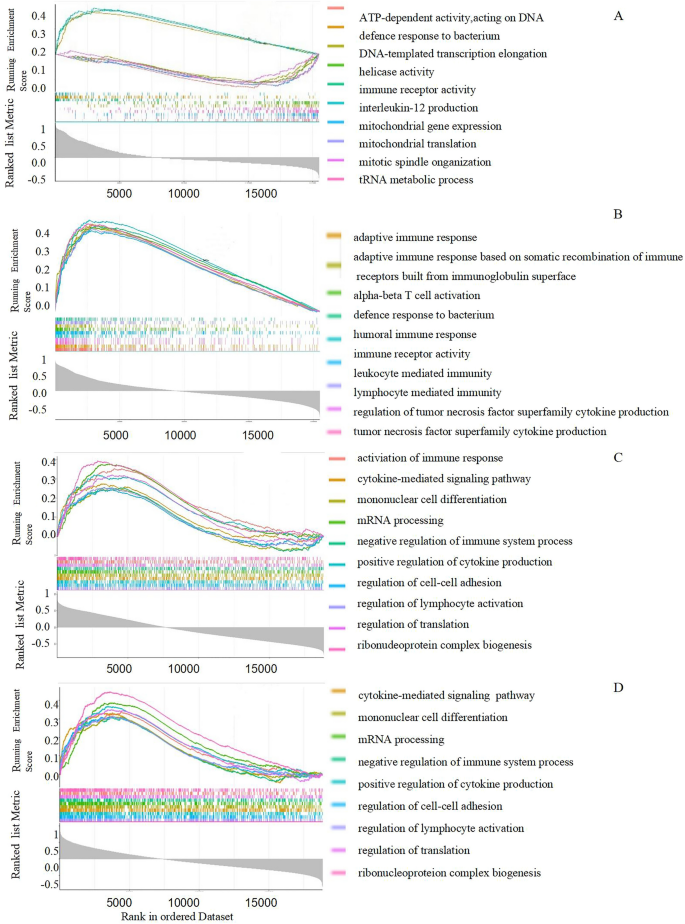 Analyse der Gensatzanreicherung. ( A , B ) Ein zusammengeführtes Anreicherungsdiagramm von SELE, CCL20 in der Kohorte GSE 147507. ( C , D ) Ein zusammengeführtes Anreicherungsdiagramm von SELE, CCL20 in der Kohorte GSE113439.
Korrelation zwischen Immuninvasion und KerngenenWir haben ssGSEA verwendet, um die möglichen Beziehungen zwischen den beiden entdeckten Hub-Genen und 28 Immunzellen zu untersuchen. SELE、CCL20 standen nachweislich in direktem Zusammenhang mit verschiedenen Immunzelltypen im GSE 147507-Datensatz (Abb.
7
A), mit Ausnahme von CD56dim-natürlichen Killerzellen, CD56bright-natürlichen Killerzellen, Gedächtnis-B-Zellen und Typ-2-T-Helferzellen. Diese beiden gefundenen Hub-Gene in GSE113439 waren mit den folgenden Zelltypen und -untergruppen verknüpft: Typ-1-T-Helferzelle, regulatorische T-Zelle, plasmazytoide dendritische Zelle, Neutrophil, natürliche Killer-T-Zelle, Mastzelle, zentrale Gedächtnis-CD4-T-Zelle, aktivierte dendritische Zelle und aktivierte CD4-T-Zelle (Abb.
7
.Abbildung 7
Analyse der Gensatzanreicherung. ( A , B ) Ein zusammengeführtes Anreicherungsdiagramm von SELE, CCL20 in der Kohorte GSE 147507. ( C , D ) Ein zusammengeführtes Anreicherungsdiagramm von SELE, CCL20 in der Kohorte GSE113439.
Korrelation zwischen Immuninvasion und KerngenenWir haben ssGSEA verwendet, um die möglichen Beziehungen zwischen den beiden entdeckten Hub-Genen und 28 Immunzellen zu untersuchen. SELE、CCL20 standen nachweislich in direktem Zusammenhang mit verschiedenen Immunzelltypen im GSE 147507-Datensatz (Abb.
7
A), mit Ausnahme von CD56dim-natürlichen Killerzellen, CD56bright-natürlichen Killerzellen, Gedächtnis-B-Zellen und Typ-2-T-Helferzellen. Diese beiden gefundenen Hub-Gene in GSE113439 waren mit den folgenden Zelltypen und -untergruppen verknüpft: Typ-1-T-Helferzelle, regulatorische T-Zelle, plasmazytoide dendritische Zelle, Neutrophil, natürliche Killer-T-Zelle, Mastzelle, zentrale Gedächtnis-CD4-T-Zelle, aktivierte dendritische Zelle und aktivierte CD4-T-Zelle (Abb.
7
.Abbildung 7
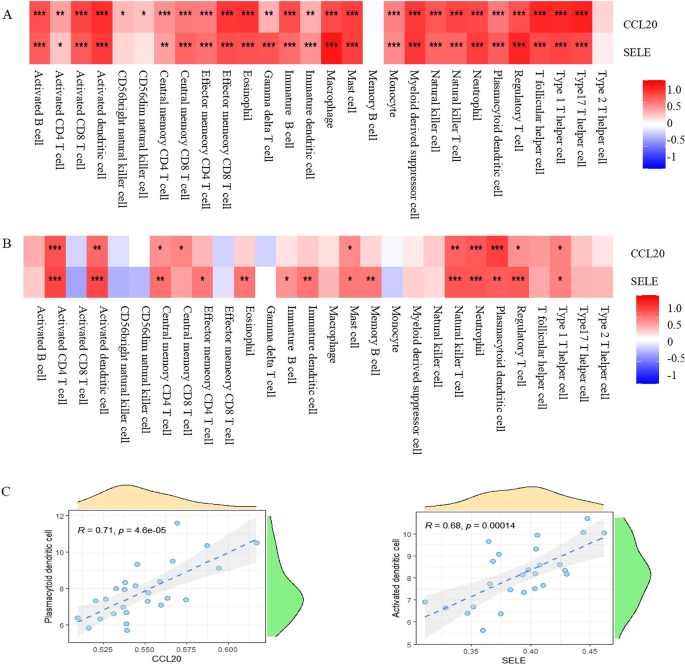 Zusammenhang zwischen den Hub-Genen und Immuninfiltration. ( A ) In der Kohorte GSE147507 korrelierten CCL20 und SELE positiv mit den meisten Zelltypen. Mit Ausnahme von CD56dim-natürlichen Killerzellen, CD56bright-natürlichen Killerzellen, Gedächtnis-B-Zellen und Typ-2-T-Helferzellen. ( In der Kohorte GSE113439 korrelierten CCL20 und SELE positiv mit vielen Zelltypen. Einschließlich: Typ-1-T-Helferzelle, regulatorische T-Zelle, plasmazytoide dendritische Zelle, Neutrophil, natürliche Killer-T-Zelle, Mastzelle, zentrale Gedächtnis-CD4-T-Zelle, aktivierte dendritische Zelle und aktivierte CD4-T-Zelle. Rot: positive Korrelation; Blau: negative Korrelation. ( C ) Laut einer Korrelationsstudie von Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Verknüpfungsgrad mit SELE aufwiesen.
Als wir die ssGSEA-Ergebnisse aus den oben genannten Datensätzen kombinierten, stellten wir fest, dass COVID-19 und PH mit der Aktivierung von CD4-T-Zellen, aktivierten dendritischen Zellen, natürlichen Killer-T-Zellen, Neutrophilen und plasmazytoiden dendritischen Zellen in Zusammenhang standen. Laut einer Korrelationsstudie nach Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Kopplungsgrad mit SELE aufwiesen (Abb.
7
C).Komodulatorische Achse TF-miRNA und TF-Gen-AssoziationenAnschließend wurde der Netzwerkanalyst verwendet (Abb.
8
A). Das erstellte Netzwerk enthielt insgesamt 19 Knoten und 18 Kanten. Es gab eine starke Korrelation zwischen Hub-Genen und TFs, und die TFs beeinflussten mehr als ein Hub-Gen im Netzwerk. Um die Verbindung zwischen miRNAs + TFs und Hub-Genen zu bewerten, erstellten wir die TF-miRNA-Komodulatorachse. Unser Netzwerk enthielt 39 Knoten, 38 Kanten und 25 miRNA-basierte Interaktionen, an denen Hub-Gene beteiligt waren, wodurch die Gesamtexpression der Hub-Gene moduliert werden konnte. In unserer Forschung wurden SELE und CCL20 als potenzielle diagnostische Biomarker für COVID-19 in Verbindung mit pulmonaler Hypertonie (PH) identifiziert. Um die Rolle dieser Gene bei der Krankheit weiter zu validieren, schlagen wir die Verwendung des FFLtools zur Analyse von Transkriptionsfaktor-Gen- und Transkriptionsfaktor-miRNA-Netzwerken vor. FFLtool ist ein Webserver, der für die Analyse von Feedforward-Loops (FFLs) entwickelt wurde und tiefe Einblicke in die Wechselwirkungen zwischen Transkriptionsfaktoren, miRNAs und Genen bietet. Durch die Einbeziehung unserer Schlüsselgene, Transkriptionsfaktoren und miRNAs in diese Analyse. Als Ergebnis erschien die FFL, die TF CCL20, miRNA miR-1256 und das Zielgen PPARG enthält, ganz oben (Abb.
8
B, ergänzende Abb.
1
). Diese FFL unter CCL20, miR-1256 und PPARG könnte ein neuartiges regulatorisches Modul bei COVID-19 sein, das durch pulmonale Hypertonie kompliziert wird.Abbildung 8
Zusammenhang zwischen den Hub-Genen und Immuninfiltration. ( A ) In der Kohorte GSE147507 korrelierten CCL20 und SELE positiv mit den meisten Zelltypen. Mit Ausnahme von CD56dim-natürlichen Killerzellen, CD56bright-natürlichen Killerzellen, Gedächtnis-B-Zellen und Typ-2-T-Helferzellen. ( In der Kohorte GSE113439 korrelierten CCL20 und SELE positiv mit vielen Zelltypen. Einschließlich: Typ-1-T-Helferzelle, regulatorische T-Zelle, plasmazytoide dendritische Zelle, Neutrophil, natürliche Killer-T-Zelle, Mastzelle, zentrale Gedächtnis-CD4-T-Zelle, aktivierte dendritische Zelle und aktivierte CD4-T-Zelle. Rot: positive Korrelation; Blau: negative Korrelation. ( C ) Laut einer Korrelationsstudie von Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Verknüpfungsgrad mit SELE aufwiesen.
Als wir die ssGSEA-Ergebnisse aus den oben genannten Datensätzen kombinierten, stellten wir fest, dass COVID-19 und PH mit der Aktivierung von CD4-T-Zellen, aktivierten dendritischen Zellen, natürlichen Killer-T-Zellen, Neutrophilen und plasmazytoiden dendritischen Zellen in Zusammenhang standen. Laut einer Korrelationsstudie nach Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Kopplungsgrad mit SELE aufwiesen (Abb.
7
C).Komodulatorische Achse TF-miRNA und TF-Gen-AssoziationenAnschließend wurde der Netzwerkanalyst verwendet (Abb.
8
A). Das erstellte Netzwerk enthielt insgesamt 19 Knoten und 18 Kanten. Es gab eine starke Korrelation zwischen Hub-Genen und TFs, und die TFs beeinflussten mehr als ein Hub-Gen im Netzwerk. Um die Verbindung zwischen miRNAs + TFs und Hub-Genen zu bewerten, erstellten wir die TF-miRNA-Komodulatorachse. Unser Netzwerk enthielt 39 Knoten, 38 Kanten und 25 miRNA-basierte Interaktionen, an denen Hub-Gene beteiligt waren, wodurch die Gesamtexpression der Hub-Gene moduliert werden konnte. In unserer Forschung wurden SELE und CCL20 als potenzielle diagnostische Biomarker für COVID-19 in Verbindung mit pulmonaler Hypertonie (PH) identifiziert. Um die Rolle dieser Gene bei der Krankheit weiter zu validieren, schlagen wir die Verwendung des FFLtools zur Analyse von Transkriptionsfaktor-Gen- und Transkriptionsfaktor-miRNA-Netzwerken vor. FFLtool ist ein Webserver, der für die Analyse von Feedforward-Loops (FFLs) entwickelt wurde und tiefe Einblicke in die Wechselwirkungen zwischen Transkriptionsfaktoren, miRNAs und Genen bietet. Durch die Einbeziehung unserer Schlüsselgene, Transkriptionsfaktoren und miRNAs in diese Analyse. Als Ergebnis erschien die FFL, die TF CCL20, miRNA miR-1256 und das Zielgen PPARG enthält, ganz oben (Abb.
8
B, ergänzende Abb.
1
). Diese FFL unter CCL20, miR-1256 und PPARG könnte ein neuartiges regulatorisches Modul bei COVID-19 sein, das durch pulmonale Hypertonie kompliziert wird.Abbildung 8
 Netzwerk für die Interaktion von TF-Genen und TF-miRNA mit gängigen DEGs. ( A ) Wir haben über die Website des Netzwerkanalysten 25 miRNAs und 11 TF-Gene vorhergesagt, die mit SELE und CCL20 interagieren. Rote Knoten: Hub-Gene; blaue Knoten: miRNA. Grüne Knoten: TF-Gene. ( B ) Wir haben mithilfe des FFL-Tools festgestellt, dass miR-1256 und PPARG unter den CCL20 möglicherweise ein neues regulatorisches Modul bei COVID-19 sind, das durch pulmonale Hypertonie kompliziert wird.
Gezielte chemische Interaktion und Identifizierung von Medikamentenkandidaten bei COVID-19 und PHGlutathion, Simvastatin, Niacin, Fenretinid, 1-Nitropyren, N -Acetyl- =0.8eml -Cystein, Nickelchlorid, Kieselsäure, Vincristin, Wasserstoffperoxid und Aflatoxin B1 gehörten zu den 10 in Betracht gezogenen Medikamenten (Ergänzende Tabelle
S5
). Diese potenziellen Medikamente interagierten mit gemeinsamen DEGs, was darauf hindeutet, dass sie zur Behandlung beider Erkrankungen eingesetzt werden könnten.Darüber hinaus wurde molekulares Docking genutzt, um die Bindungsmechanismen zwischen den Medikamenten-Hub-Genen vorherzusagen (Ergänzende Tabelle
S6
). Abbildung
9
zeigt die Ergebnisse der molekularen Dockinganalyse. Es wurde festgestellt, dass die Bindungsstellen von FENRETINID, 1-NITROPYREN und AFLATOXIN B1 an die beiden Zielproteine geringere Stabilisierungsenergien aufweisen. Daher könnten diese drei in Betracht gezogenen Medikamente eines Tages zur Behandlung sowohl von COVID-19 als auch von PH eingesetzt werden (Abb.
9
). Anschließend führten wir auch Arzneimittelvorhersagen für diese beiden Schlüsselgene in der CTD-Datenbank durch und fanden heraus, dass Lipopolysaccharide, Bisphenol A, Acetaminophen, Benzo(a)pyren, Siliziumdioxid, Tetrachlordibenzodioxin, Titandioxid, 1-Nitropyren, 2,2′,4,4′-Tetrabromdiphenylether und 2-Anisidin (Ergänzende Tabelle
S7
) vorhanden sind. Durch die Kombination der Enrichr- und CTD-Datenbanken ist es nicht schwierig herauszufinden, dass 1-NITROPYREN in beiden Datenbanken vorhergesagt werden kann. Daher führten wir zusätzlich molekulardynamische Simulationen durch.Abbildung 9
Netzwerk für die Interaktion von TF-Genen und TF-miRNA mit gängigen DEGs. ( A ) Wir haben über die Website des Netzwerkanalysten 25 miRNAs und 11 TF-Gene vorhergesagt, die mit SELE und CCL20 interagieren. Rote Knoten: Hub-Gene; blaue Knoten: miRNA. Grüne Knoten: TF-Gene. ( B ) Wir haben mithilfe des FFL-Tools festgestellt, dass miR-1256 und PPARG unter den CCL20 möglicherweise ein neues regulatorisches Modul bei COVID-19 sind, das durch pulmonale Hypertonie kompliziert wird.
Gezielte chemische Interaktion und Identifizierung von Medikamentenkandidaten bei COVID-19 und PHGlutathion, Simvastatin, Niacin, Fenretinid, 1-Nitropyren, N -Acetyl- =0.8eml -Cystein, Nickelchlorid, Kieselsäure, Vincristin, Wasserstoffperoxid und Aflatoxin B1 gehörten zu den 10 in Betracht gezogenen Medikamenten (Ergänzende Tabelle
S5
). Diese potenziellen Medikamente interagierten mit gemeinsamen DEGs, was darauf hindeutet, dass sie zur Behandlung beider Erkrankungen eingesetzt werden könnten.Darüber hinaus wurde molekulares Docking genutzt, um die Bindungsmechanismen zwischen den Medikamenten-Hub-Genen vorherzusagen (Ergänzende Tabelle
S6
). Abbildung
9
zeigt die Ergebnisse der molekularen Dockinganalyse. Es wurde festgestellt, dass die Bindungsstellen von FENRETINID, 1-NITROPYREN und AFLATOXIN B1 an die beiden Zielproteine geringere Stabilisierungsenergien aufweisen. Daher könnten diese drei in Betracht gezogenen Medikamente eines Tages zur Behandlung sowohl von COVID-19 als auch von PH eingesetzt werden (Abb.
9
). Anschließend führten wir auch Arzneimittelvorhersagen für diese beiden Schlüsselgene in der CTD-Datenbank durch und fanden heraus, dass Lipopolysaccharide, Bisphenol A, Acetaminophen, Benzo(a)pyren, Siliziumdioxid, Tetrachlordibenzodioxin, Titandioxid, 1-Nitropyren, 2,2′,4,4′-Tetrabromdiphenylether und 2-Anisidin (Ergänzende Tabelle
S7
) vorhanden sind. Durch die Kombination der Enrichr- und CTD-Datenbanken ist es nicht schwierig herauszufinden, dass 1-NITROPYREN in beiden Datenbanken vorhergesagt werden kann. Daher führten wir zusätzlich molekulardynamische Simulationen durch.Abbildung 9
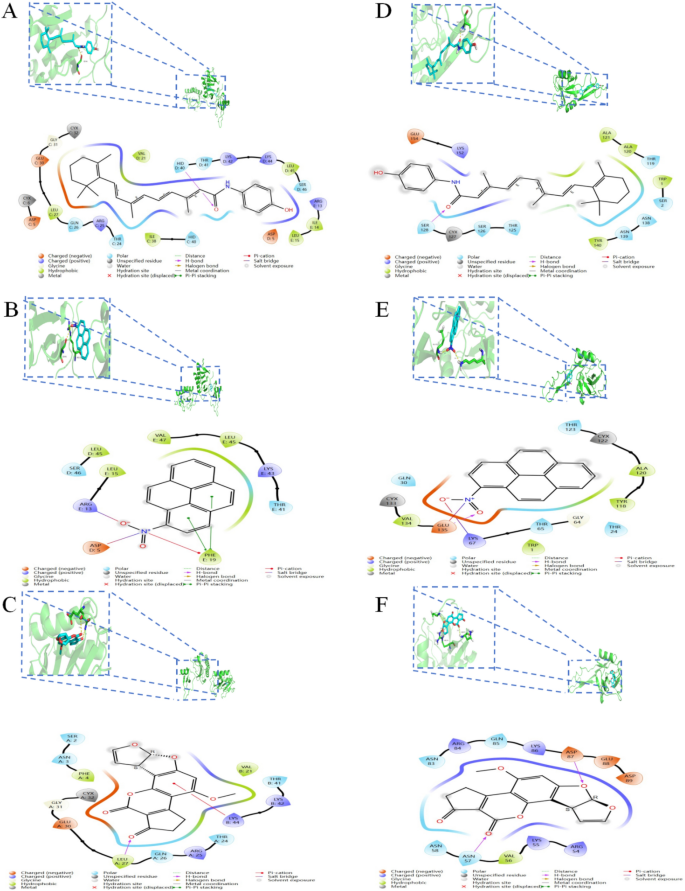 3D- und 2D-Dockingmuster für molekulare Verbindungen für ( A ) FENRETINID, ( B ) 1-NITROPYREN, ( C ) AFLATOXIN B1 mit CCL20 bzw. ( D ) FENRETINID, ( E ) 1-NITROPYREN, ( F ) AFLATOXIN B1 mit SELE.
Molekulardynamische Simulation des 1-Nitropyren-CCL20/SELE-KomplexesBasierend auf den Ergebnissen des molekularen Dockings wurden 100-ns-MD-Simulationen des 1-Nitropyren-CCL20/SELE-Komplexes durchgeführt, um die durch das molekulare Docking erzielten dynamischen Eigenschaften zu untersuchen. Die Endergebnisse umfassten die mittlere quadratische Abweichung (RMSD), die mittlere quadratische Fluktuation (RMSF), den Trägheitsradius (Rg), die lösungszugängliche Oberfläche (SASA) und die Änderungen der Wasserstoffbrücken während des gesamten 100-ns-Simulationsprozesses.Die RMSD-Kurven des 1-Nitropyren-CCL20-Komplexes näherten sich nach 28 ns allmählich dem Gleichgewicht, mit einer RMSD-Schwankung von 0,25–0,3 nm. Darüber hinaus näherten sich alle RMSD-Kurven des 1-Nitropyren-SELE-Komplexes nach 60–88 ns dem Gleichgewicht, und die mittlere RMSD betrug weniger als 0,35 nm (Abb.
10
A). Die Rg-Kurven der 1-Nitropyren-CCL20- und 1-Nitropyren-SELE-Komplexe blieben während des gesamten Prozesses stabil, mit durchschnittlichen Rg-Werten von 1,88 nm bzw. 1,68 nm (Abb.
10
. Die niedrigen Rg-Werte weisen darauf hin, dass das Protein während des Simulationsprozesses von 100 ns kompakt blieb. Im 1-Nitropyren-CCL20-Komplex schwankte der RMSF der C-Kette des CCL20-Proteins erheblich; dies war besonders im Bereich der Aminosäuren 27–36 zu beobachten, der, wie aus der Proteinstruktur ersichtlich, in der Schleifenregion der C-Kette liegt, was zu einer größeren Flexibilität während des 100-ns-Prozesses führt. Die RMSF-Werte der α-Helix und der β-Faltung in der C-Kette blieben während des Simulationsprozesses relativ niedrig, während die RMSF-Werte der A- und B-Ketten sich im Wesentlichen gleichzeitig änderten, was darauf hindeutet, dass das kleine 1-Nitropyren-Molekül wenig Einfluss auf die Gesamtflexibilität des CCL20-Proteins hatte. Im 1-Nitropyren-SELE-Komplex wurden große RMSF-Werte im Bereich der 124–140 Aminosäuren beobachtet, der laut molekularem Docking die Bindungstasche des kleinen Moleküls umgibt. Daher spekulieren wir, dass die Zugabe von 1-Nitropyren die Flexibilität der Aminosäuren an der Bindungsstelle erhöhte. Die Ergebnisse zeigen einen großen RMSF-Wert (Abb.
10
C,D). Im 100-ns-Simulationsprozess blieb die Anzahl der Wasserstoffbrücken im 1-Nitropyren-CCL20/SELE-Komplex bei 2, während die maximale Anzahl von Wasserstoffbrücken im 1-Nitropyren-CCL20-Komplex 3 betrug. Der 1-Nitropyren-SELE-Komplex könnte bis zu fünf Wasserstoffbrücken aufweisen. Diese beträchtliche Anzahl von Wasserstoffbrücken trägt dazu bei, die Stabilität des Komplexes aufrechtzuerhalten (Abb.
10
E). Die SASA-Kurven der 1-Nitropyren-CCL20- und 1-Nitropyren-SELE-Komplexe blieben während des gesamten Prozesses stabil und die mittleren SASA-Werte waren wie folgt: Die niedrigen SASA-Werte von 128 nm 2 und 92 nm 2 (Abb.
10
F).Abbildung 10
3D- und 2D-Dockingmuster für molekulare Verbindungen für ( A ) FENRETINID, ( B ) 1-NITROPYREN, ( C ) AFLATOXIN B1 mit CCL20 bzw. ( D ) FENRETINID, ( E ) 1-NITROPYREN, ( F ) AFLATOXIN B1 mit SELE.
Molekulardynamische Simulation des 1-Nitropyren-CCL20/SELE-KomplexesBasierend auf den Ergebnissen des molekularen Dockings wurden 100-ns-MD-Simulationen des 1-Nitropyren-CCL20/SELE-Komplexes durchgeführt, um die durch das molekulare Docking erzielten dynamischen Eigenschaften zu untersuchen. Die Endergebnisse umfassten die mittlere quadratische Abweichung (RMSD), die mittlere quadratische Fluktuation (RMSF), den Trägheitsradius (Rg), die lösungszugängliche Oberfläche (SASA) und die Änderungen der Wasserstoffbrücken während des gesamten 100-ns-Simulationsprozesses.Die RMSD-Kurven des 1-Nitropyren-CCL20-Komplexes näherten sich nach 28 ns allmählich dem Gleichgewicht, mit einer RMSD-Schwankung von 0,25–0,3 nm. Darüber hinaus näherten sich alle RMSD-Kurven des 1-Nitropyren-SELE-Komplexes nach 60–88 ns dem Gleichgewicht, und die mittlere RMSD betrug weniger als 0,35 nm (Abb.
10
A). Die Rg-Kurven der 1-Nitropyren-CCL20- und 1-Nitropyren-SELE-Komplexe blieben während des gesamten Prozesses stabil, mit durchschnittlichen Rg-Werten von 1,88 nm bzw. 1,68 nm (Abb.
10
. Die niedrigen Rg-Werte weisen darauf hin, dass das Protein während des Simulationsprozesses von 100 ns kompakt blieb. Im 1-Nitropyren-CCL20-Komplex schwankte der RMSF der C-Kette des CCL20-Proteins erheblich; dies war besonders im Bereich der Aminosäuren 27–36 zu beobachten, der, wie aus der Proteinstruktur ersichtlich, in der Schleifenregion der C-Kette liegt, was zu einer größeren Flexibilität während des 100-ns-Prozesses führt. Die RMSF-Werte der α-Helix und der β-Faltung in der C-Kette blieben während des Simulationsprozesses relativ niedrig, während die RMSF-Werte der A- und B-Ketten sich im Wesentlichen gleichzeitig änderten, was darauf hindeutet, dass das kleine 1-Nitropyren-Molekül wenig Einfluss auf die Gesamtflexibilität des CCL20-Proteins hatte. Im 1-Nitropyren-SELE-Komplex wurden große RMSF-Werte im Bereich der 124–140 Aminosäuren beobachtet, der laut molekularem Docking die Bindungstasche des kleinen Moleküls umgibt. Daher spekulieren wir, dass die Zugabe von 1-Nitropyren die Flexibilität der Aminosäuren an der Bindungsstelle erhöhte. Die Ergebnisse zeigen einen großen RMSF-Wert (Abb.
10
C,D). Im 100-ns-Simulationsprozess blieb die Anzahl der Wasserstoffbrücken im 1-Nitropyren-CCL20/SELE-Komplex bei 2, während die maximale Anzahl von Wasserstoffbrücken im 1-Nitropyren-CCL20-Komplex 3 betrug. Der 1-Nitropyren-SELE-Komplex könnte bis zu fünf Wasserstoffbrücken aufweisen. Diese beträchtliche Anzahl von Wasserstoffbrücken trägt dazu bei, die Stabilität des Komplexes aufrechtzuerhalten (Abb.
10
E). Die SASA-Kurven der 1-Nitropyren-CCL20- und 1-Nitropyren-SELE-Komplexe blieben während des gesamten Prozesses stabil und die mittleren SASA-Werte waren wie folgt: Die niedrigen SASA-Werte von 128 nm 2 und 92 nm 2 (Abb.
10
F).Abbildung 10
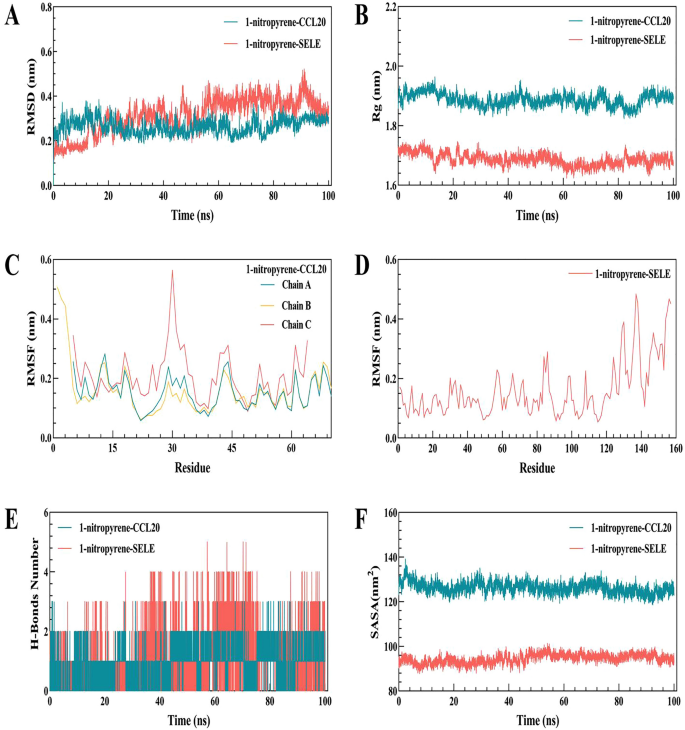 100 ns Moleküldynamik-Simulationsanalyse des 1-Nitropyren-CCL20/SELE-Komplexes. ( A ) RMSD-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes, ( B ) Rg-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes, ( C ) RMSF-Kurve des 1-Nitropyren-CCL20-Komplexes, ( D ) RMSF-Kurve des 1-Nitropyren-SELE-Komplexes, ( E ) Wasserstoffbrückenänderungskurve des 1-Nitropyren-CCL20/SELE-Komplexes und ( F ) SASA-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes.
DiskussionNach der ersten Beobachtung von Xie und Kollegen
12
, dass COVID-19 mit einem Anstieg von Herz-Kreislauf-Erkrankungen einhergeht, haben zahlreiche klinische Studien und Metaanalysen eine erhöhte Inzidenz von akuten Koronarsyndromen, Myokarditis, Perikarditis, Herzinsuffizienz und Arrhythmien bestätigt
13
,
14
,
15
,
16
,
17
,
18
,
19
,
20.
Zu den Folgen einer schweren COVID-19-Erkrankung gehören systemische Hypoxie, akutes Atemnotsyndrom, Hyperkoagulation, Sepsis, Entzündungen, metabolischer Stress und Zytokinstürme, die alle das Herz-Kreislauf-System belasten und schließlich zu einer Blutdruckentgleisung führen können
21
,
22
,
23
,
24
,
25
.Pulmonale Hypertonie als schwerwiegende kardiopulmonale Komplikation von COVID-19 erhöht die Wahrscheinlichkeit, dass eine Behandlung auf der Intensivstation, künstliche Beatmung, extrakorporale Membranoxygenierung (ECMO) oder sogar der Tod erforderlich werden. Daher könnte das frühzeitige Erkennen von hohem Pulmonalarteriendruck bei SARS-CoV-2-Patienten die Langzeitprognose der Patienten verbessern und die Hospitalisierungsrate sowie die Sterberate aufgrund solcher Komplikationen minimieren
7
,
8
,
9
,
26.
De Jong CMM
27
fand jedoch heraus, dass chronische thromboembolische pulmonale Hypertonie nach einer COVID-19-assoziierten Lungenembolie keine häufigere Langzeitkomplikation ist als nach einer nicht-COVID-19-assoziierten Lungenembolie. Unabhängig davon, ob dieses Phänomen auf große Unterschiede in der Pathogenese zurückzuführen ist, ist es besonders wichtig, die Pathogenese von COVID-19 in Kombination mit pulmonaler Hypertonie zu untersuchen.In dieser Studie wurde versucht, Moleküle zu untersuchen, die mit der Pathogenese von COVID in Kombination mit PH in Zusammenhang stehen, und zwar mithilfe verschiedener bioinformatischer Methoden. Anschließend wurde durch die Bewertung signifikanter Marker ein umfassendes Diagnosemuster erstellt. Die Expression jedes Gens wurde quantifiziert und bewertet, wobei höhere Werte mit einem größeren Vorhersagepotenzial verbunden waren. Diese Vorhersagewerte könnten zur Überwachung und Frühintervention bei COVID-19-Patienten, insbesondere bei Patienten mit PH, verwendet werden. Ein weiteres Ziel war die Identifizierung spezifischer Medikamente, die auf Schlüsselgene abzielen, die mit COVID-19 in Verbindung stehen und mit PH einhergehen, und so sowohl die Diagnose als auch die Behandlung dieser Erkrankung verbessern könnten.Ähnlich wie bei den genetischen Ursachen der pulmonal-arteriellen Hypertonie kann der Schweregrad von COVID-19 durch Variationen in denselben Genen beeinflusst werden. Viele der pathobiologischen Kennzeichen der pulmonal-arteriellen Hypertonie sind auch bei der durch COVID-19 induzierten pulmonalen Vaskulopathie vorhanden, darunter Endodermatitis, Vaskulitis-mediale Hypertrophie und Proliferation glatter Muskelzellen. Insbesondere eine endotheliale Dysfunktion ist ein häufiges Merkmal der bei COVID-19-Patienten beobachteten klinischen Manifestationen. Das Coronavirus SARS-CoV-2 gelangt in die Wirtszellen, indem es sein Spike-Glykoprotein an das Angiotensin-konvertierende Enzym 2 (ACE2), den Sialinsäurerezeptor, die transmembranöse Serinprotease 2 (TMPRSS2) und den Induktor der extrazellulären Matrix-Metalloproteinase (CD147) bindet; Catepsin B und L sind ebenfalls am Eindringen des Virus beteiligt. Alle diese Faktoren werden in Endothelzellen exprimiert
28
,
29
,
30
,
31
Überaktivierte Thrombozyten verursachen Zytokinstürme und Thrombosen. Studien haben gezeigt, dass Thrombozyten, die entzündungsfördernde Moleküle exprimieren und virale RNA tragen, besonders wahrscheinlich hochaktiv sind
32
.Unter den C-DEGs fanden wir beim Vergleich der COVID-19- und PH-Datensätze 47 stark exprimierte Gene und 15 schwach exprimierte Gene. Mithilfe der GO-Analyse stellten wir fest, dass C-DEGs mit den folgenden Begriffen angereichert waren: Entzündungsreaktion, immunologische Reaktion, Zelloberflächenrezeptor, zelluläre Reaktion auf Interferon-gamma, positive Regulierung von NF-kB und Reaktion auf Lipopolysaccharide. TNF-Signalweg, Infektion mit Herpes-simplex-Virus 1, virale Proteininteraktion mit Zytokin und Zytokinrezeptor, Malaria, Lipid und Atherosklerose, PPAR-Signalweg und PPAR-Signalweg waren alle unter den C-DEGs in der KEGG-Anreicherungsanalyse angereichert. Diese Ergebnisse legen stark nahe, dass eine Immunentzündung eine treibende Kraft bei der Entstehung und dem Fortschreiten von COVID-19 in Verbindung mit PH ist. Eine Infektion mit SARS-CoV-2 kann einen Zytokinsturm auslösen, der zu systemischer Entzündung und vaskulärer Endothelzellschädigung führt. Diese Veränderungen können Hyperkoagulabilität und intravaskuläre Thrombose verursachen und gleichzeitig den pulmonalen Gefäßwiderstand erhöhen. Diffuse Mikroangiopathie und Mikrothrombose, die durch eine umfassende Beeinträchtigung der vaskulären Endothelfunktion verursacht werden, können das Ungleichgewicht des Lungenventilations-/Blutflussverhältnisses weiter verschlimmern, und eine Erhöhung des pulmonalen Rechts-Links-Shunts kann die Hypoxie weiter verschlimmern und so die pulmonale Gefäßkontraktion und -umgestaltung fördern. Wenn der Körper von einem Virus angegriffen wird, kann dieser außerdem T-Zellen aktivieren und die Überexpression von IFN-induzierten Genen fördern, die ebenfalls zur Apoptose von Endothelzellen und damit zu PH führen können. Dies hat viele Gemeinsamkeiten mit den Ergebnissen der Studie von LuisG
33
.Um unter den 62 identifizierten DEGs weiter nach den Kerngenen zu suchen, die mit COVID-19 in Kombination mit PH in Verbindung stehen, verwendeten wir maschinelles Lernen, um potenzielle diagnostische Biomarker auszuwählen. Die LASSO-Regressionsanalyse identifizierte 20 Gene mit der geringsten binominalen Abweichung. Der Random-Forest-Ansatz identifizierte 10 Kandidaten, nachdem die DEGs nach Signifikanz eingestuft wurden, und die SVM-RFE-Methode identifizierte 8 Gene mit dem geringsten Fehler und der besten Genauigkeit nach 100-fachen Änderungen. Anhand der Schnittmenge der Ergebnisse der drei Algorithmen wurden SELE und CCL20 als Kerngene identifiziert. Es stellte sich heraus, dass diese beiden Schlüsselgene im Validierungssatz im Vergleich zueinander statistisch signifikante Unterschiede aufwiesen. Als Nächstes erstellten wir ein Vorhersagemodell, um noch fundiertere Vorhersagen zu COVID-19 in Kombination mit PH zu treffen, indem wir die Werte in der Tabelle auswerteten, wobei höhere Werte eine größere Wahrscheinlichkeit darstellten, nach COVID-19 an PH zu erkranken. Durch Zeichnen der ROC-, PR- und DCA-Kurven wurde festgestellt, dass das Vorhersagemodell zuverlässig war.Eines der identifizierten Kerngene war SELE. Die Aktivierung von Endothelzellen durch Zytokine führt zur Expression eines Zelloberflächen-Glykoproteins namens SELE, das die Anhaftung zirkulierender Monozyten und Lymphozyten an Endothelzellen erleichtert
34.
Der Plasmamarker für endotheliale Dysfunktion oder Verletzung ist lösliches E-Selectin (sE-Selectin), das von beschädigten oder dysfunktionalen Endothelzellen abgesondert wird
35
,
36.
Der Membrantransport von IFN-gamma-R2 zum Golgi-Apparat und die ordnungsgemäße IFN-gamma-R-Zusammensetzung erfordern beide die Beteiligung von E-Selectin. Die Aktivierung der BTK-Kinase wird durch die Interaktion eines E-selektiven Proteins ausgelöst, das wiederum einen funktionellen IFN-gamma-R bildet, der an einen anderen funktionellen IFN-gamma-R binden kann und so eine effiziente angeborene Reaktion von Makrophagen auf eine intrazelluläre bakterielle Infektion erzeugt
37
,
38
. Die Bedeutung von Endothelzellen bei der Verbreitung von SARS-CoV-2 wird zunehmend akzeptiert. Wahrscheinlich ist die Funktion von E-Selectin bei der Chemotaxis von Leukozyten während einer Entzündung der grundlegende Mechanismus, der hier am Werk ist. Die Expression von E-Selectin auf der Oberfläche nimmt zu, was den Eintritt von Leukozyten in das Gewebe und die Einleitung einer Entzündung zur Bekämpfung der Infektion erleichtern könnte
39
,
40.
DM Smadja et al. entdeckten, dass PH an zirkulierende Endothelzellen, lösliches E-Selectin und sVCAM gekoppelt ist, nicht jedoch an endotheliale Vorläuferzellen, CD34(+)CD133(+)-Zellen oder den vaskulären endothelialen Wachstumsfaktor (VEGF)
41.
Ähnlichkeiten zwischen diesen und den Ergebnissen unserer Studie legen nahe, dass SELE ein Schlüsselmolekül der pathogenen COVID-19-gekoppelten PH sein könnte.CCL20, auch bekannt als Makrophagen-Entzündungsprotein-3α oder durch Leberaktivierung reguliertes Chemokin, ist ein weiteres Gen von zentraler Bedeutung, das wir untersucht haben. CCL20 ist ein CC-Chemokin, das spezifisch mit CCR6 interagiert. Neben der Rekrutierung unreifer dendritischer Zellen, Effektor-/Gedächtnis-T-Zellen und B-Zellen spielt dieses Chemokin auch eine entzündungsfördernde Rolle bei der Aufrechterhaltung der Homöostase. Es spielt eine entscheidende Rolle bei der Aufrechterhaltung des regelmäßigen Verkehrs von Immunzellen und beim Auslösen einer T-Zell-abhängigen Entzündung. CCL20 reguliert das richtige Maß an Entzündung, indem es ein feines Gleichgewicht zwischen offensiver und defensiver Immunität aufrechterhält
42
,
43
,
44
,
45
. Es rekrutiert auch Th17-Zellen und regulatorische T-Zellen an Entzündungsherde, da CCR6 auf diesen Zelltypen vorhanden ist. Bei Patienten mit COVID-19 wurden sowohl in der bronchoalveolären Lavage (BAL)-Flüssigkeit als auch in Plasmaproben erhöhte CCL20-Werte festgestellt
46
. Pulmonalarterielle Hypertonie bei Personen mit SSc ist mit erhöhten CCL20-Serumspiegeln assoziiert
44
. Allerdings gibt es bisher nur wenig Forschung zu CCL20 bei COVID- und PH-Patienten.Die funktionelle Anreicherung betraf in erster Linie die adaptive Immunantwort, die durch Leukozyten und Lymphozyten vermittelte Immunantwort und die durch Zytokine wie IL-12 und TNF-a vermittelte entzündungsfördernde Reaktion, die die Proliferation von glatten Muskelzellen der Lungenarterie förderte und eine vaskuläre Umgestaltung herbeiführte, wie durch GSEA von SELE und CCL20 in den Datensätzen von COVID-19 und PH bestimmt wurde.Mithilfe einer ssGSEA-Analyse untersuchten wir die Beziehung zwischen SELE und CCL20 sowie 23 verschiedenen Arten von Immunzellen in den COVID-19- und PH-Datensätzen. Dabei stellten wir fest, dass aktivierte CD4-T-Zellen, aktivierte dendritische Zellen, natürliche Killer-T-Zellen, Neutrophile und plasmazytoide dendritische Zellen alle mit COVID-19 und PH in Verbindung standen. Die höchste Pearson-Korrelation wurde zwischen CCL20 und plasmazytoiden dendritischen Zellen und die höchste Verbindung zwischen SELE und aktivierten dendritischen Zellen festgestellt. Die immunologische Reaktion verschiedener Zellen kann durch SELE und CCL20 reguliert werden, was zu einer erhöhten Inzidenz von COVID-19-gekoppelter PH führt. Während einer akuten Coronavirus-Infektion werden Lymphozyten, einschließlich NK-Zellen, aktiviert und wandern in die Lunge, da sich dort entzündliche mononukleäre Makrophagen und Neutrophile ansammeln, die Zytokine und Chemokine freisetzen. Wenn die SELE- und CCL20-Werte sinken, nehmen Anzahl und Funktion der NK-Zellen ab. PH entwickelte sich infolge einer erhöhten Empfindlichkeit gegenüber COVID-19 und einer Veränderung der Lungenarterienwände durch die Zerstörung von NK-Zellen. Eine chronische Gewebeentzündung kann durch eine CD4+T-Zell-vermittelte zelluläre Immunität hervorgerufen werden, eine besondere zelluläre Immunantwort, die von CD4+T-Zellen gesteuert wird
47
,
48
,
49
,
50.
Es kommt zu einer Invasion von Lymphozyten (meistens T-Zellen) und mononukleären phagozytischen Zelllinien, was zu einer exsudativen Entzündung führt. Die Pathophysiologie der durch COVID-19 komplizierten PH wird auch durch SELE- und CCL20-vermittelte Neutrophilenproliferation beeinflusst. Durch die Sekretion von extrazellulären Fallen von Neutrophilen, die wiederum die Schädigung von Lungenarterien-Endothelzellen und die Proliferation von glatten Muskelzellen erhöhen, tragen Neutrophile dazu bei, die Entzündung sowohl aufrechtzuerhalten als auch zu verschlimmern.Laut einer Pearson-Analyse zeigten SELE, CCL20 und dendritische Zellen die größte Korrelation unter den fünf Zelltypen. Laut einer Forschung von Tomonari Sumi et al.
51
nahmen dendritische Zellen bei Personen mit COVID-19-Folgeerscheinungen dramatisch ab . Etwa 7 Monate nach der SARS-CoV-2-Infektion entdeckten Perez-Gomez A et al.
52
, dass die Zahl der dendritischen Zellen in vivo erheblich abnahm. Zwischen dieser Schlussfolgerung und unserer eigenen bestehen Parallelen. Bei Menschen mit COVID-19, die auch an PH leiden, ist die Zahl reifer myeloider DC und der damit verbundenen Funktionsstörungen zurückgegangen. Daher sind DC-Zellen nicht in der Lage, eine Immunreaktion einzuleiten, indem sie die Aktivierung und Vermehrung primärer T-Zellen fördern, um den Körper vor virusbedingten Schäden zu schützen.Neben der Diagnose ist auch die Identifizierung von Medikamenten, die auf die Pathogenese abzielen, eine wichtige Richtung für uns. Die derzeitige Behandlung von COVID-19 hängt in erster Linie von unterstützender Pflege sowie dem Einsatz antiviraler und immunmodulatorischer Medikamente ab. Angesichts der Verteilung der Bevölkerung mit Komorbiditäten, insbesondere der überwiegend mittleren und älteren Bevölkerungsgruppe, könnten Polypharmazeutika und Arzneimittelwechselwirkungen offensichtlich sein. Leider ist das potenzielle Risiko von Arzneimittelwechselwirkungen weitgehend unbekannt, da die meisten Studien zu COVID-19 keine Details zu Wechselwirkungen zwischen im Rahmen der COVID-19-Behandlung verwendeten Medikamenten und Komedikamenten zur Behandlung anderer Komorbiditäten bei diesen Patienten liefern
53
. Darüber hinaus kann die Verwendung einiger häufig verwendeter Medikamente bei COVID-Patienten zu einer erhöhten Anzahl unerwünschter Lungeneffekte führen
54
. Die derzeitige Richtung besteht darin, geeignete Medikamente zu finden, die eine bestimmte therapeutische Wirkung und relativ wenige Nebenwirkungen haben. Aufgrund ihrer Bedeutung für die Pathogenese von COVID-19 und PH haben wir in dieser Studie SELE und CCL20 als Ziele für die Arzneimittelvorhersage und das molekulare Docking ausgewählt. Die Bindungsenergie dieser beiden Moleküle mit FENRETINID, 1-NITROPYREN und AFLATOXIN B1 war die niedrigste aller acht erwarteten Medikamente, was nicht erwartet wurde. Derzeit gibt es einige Berichte, dass 1-NITROPYREN und AFLATOXIN B1 die Symptome von COVID-19 und PH lindern können
55
,
56
,
57
,
58
,
59
,
60
,
61
,
62.
Auch als Nitroverbindung fanden WenXia Feng et al. heraus, dass die Behandlung mit inhaliertem Stickoxid bei der Verringerung und Stabilisierung von PASP hilfreich war und auch das Risiko einer Rechtsherzinsuffizienz bei COVID-19 mit pulmonaler Hypertonie verringern könnte
63
. In dieser Studie stellten wir fest, dass wir durch die Durchführung von Medikamentenvorhersagen für die Schlüsselgene, die COVID mit PH kombiniert, sowohl in der Enricher-Datenbank als auch in der CTD-Datenbank vorhersagen konnten, dass 1-NITROPYREN ein potenzielles therapeutisches Medikament ist, und dass wir durch molekulares Docking und molekulare Dynamiksimulationen herausfanden, dass die Kombination von 1-NITROPYREN und den beiden Schlüsselgenen äußerst stabil war. Daher hat es großes Potenzial, als Medikament zur Behandlung dieser Komplikation eingesetzt zu werden. Aufgrund seiner positiven Auswirkungen auf die Glukosetoleranz, den Lipidspiegel und den Körperfettanteil wurde das synthetische Retinid-Derivat Fenretinid für eine Vielzahl von medizinischen Zwecken eingesetzt, darunter zur Krebsprävention und -behandlung, zur Verbesserung der Arteriosklerose und zur Linderung der nichtalkoholischen Fettlebererkrankung. Seine Fähigkeit, die Produktion von Entzündungsmediatoren zu reduzieren und die Makrophagenpolarisation zu verhindern, könnte hier der primäre Mechanismus sein
63
,
64
,
65.
Fenretinid hemmt die Freisetzung entzündungsfördernder Faktoren (IL-1β, MCP1, iNOS und TNF-α). Fenretinid könnte die NF-κB-Signalgebung hemmen, indem es die nukleare Translokation des Proteins durch Herunterregulierung der IKKβ- und IκBα-Phosphorylierung verringert
64.
Darüber hinaus ist die verzögerte Freisetzung von IFN-I bei einer SARS-CoV-Infektion als Mechanismus bekannt, der die antivirale Reaktion des Körpers verlangsamt. Die mit der Umgehung von IFN-I verbundenen viralen Mechanismen sind vielschichtig und umfassen die Sequestrierung und Abschirmung von RNA in Doppelmembranvesikeln, die Modifikation viraler mRNA-5-Cap-Strukturen und das spezifische Angreifen antiviraler zellulärer Signalwege. Bei SARS-CoV und MERS-CoV wirkt die IFN-I-Produktion nur in den frühen Stadien nach der Infektion schützend; zu späteren Zeitpunkten hingegen, wenn die Immunantwort verstärkt ist, werden IFN-I und inflammatorische Zytokine pathogen
66
,
67
,
68
,
69
. Sowohl beim Zika-Virus als auch beim Dengue-Virus hemmte Fenretinid das nichtstrukturelle Protein 5 (NS5), das zur Virulenz beitrug, indem es die Produktion von IFN-I verhinderte
70
. Daher ist es möglich, dass Fenretinid auch die Mechanismen beeinflusst, die die IFN-I-Umgehung bei Coronavirus-Infektionen regulieren. Es gibt jedoch noch wenige relevante Erkenntnisse und der genaue Wirkmechanismus ist unklar, sodass eingehendere pharmakologische Studien erforderlich sind.Unsere Literaturrecherche ergab, dass es an Forschung zum gemeinsamen Mechanismus von COVID-19 und PH mangelt, insbesondere an bioinformatischen Studien. Hier suchten wir nach C-DEGs, testeten Core-Gene mit einem maschinellen Algorithmus und erstellten ein Modell zur Vorhersage von COVID-19 in Kombination mit PH. Wir untersuchten, wie diese beiden wesentlichen Gene mit Krankheiten in Verbindung stehen, und machten Vorhersagen über die Transkriptionsfaktoren und miRNAs, die sie regulieren. Am Ende nutzten wir molekulares Docking und Targeting-Vorhersagen für zwei wichtige Gene, um zu bestimmen, welche Medikamente am wirksamsten wären.Unsere Forschung wies jedoch gewisse Lücken auf. Zunächst stellten wir durch molekulares Docking fest, dass Fenretinid, ein zielgerichtetes Medikament für SELE und CCL20, ein neues Ziel für die Behandlung der kombinierten PH mit COVID 19 sein könnte, obwohl der Wirkmechanismus noch weiter untersucht werden muss. Zur weiteren Verifizierung der aktuellen Ergebnisse ist eine externe Validierung erforderlich. Darüber hinaus ist eine In-vitro-Modellvalidierung erforderlich, um die Kerngenfunktionen zusätzlich zu erforschen.Materialen und MethodenDatenerfassungWir haben die gesamte GEO-Datenbank (
www.ncbi.nlm.nih.gov/geo/
) nach den Begriffen „COVID-19“ und „Pulmonale Hypertonie“
71
durchsucht . Alle sequenzierten Daten stammten von Menschen, und alle gescreenten Datensätze umfassten sowohl Kontrollpersonen als auch erkrankte Personen. Die Datensätze GSE113439, GSE147507 und GSE53408 haben einen rigorosen Screeningprozess durchlaufen und wurden schließlich ausgewählt. GSE147507 enthielt insgesamt 23 Patienten mit COVID-19 und Proben von 55 gesunden Freiwilligen, während GSE113439 15 Patienten mit PH und 11 normale Kontrollpersonen umfasste. Außerdem gab es im Datensatz GSE53408 11 gesunde Personen und 12 mit sehr schwerer PH, sowie 9 gesunde Personen und 26 mit COVID-19 im Datensatz GSE196822. Eine Übersicht dieser Datensätze finden Sie in Tabelle
S8
. Das genomische Profil der DEGs wurde log2-transformiert und Gensymbole wurden mithilfe von Annotationsdokumentationen aus relevanten Datensätzen mit Sonden abgeglichen. Schließlich wurde eine Genmatrix zur weiteren Analyse extrahiert, wobei Gensymbole in Spalten und Probennamen in Zeilen dargestellt wurden.DEG-ScreeningDEGs wurden zwischen PH und Kontrollen im GSE113439-Datensatz mithilfe der Pakete Limma und GEOquery im R-Programm sowie zwischen COVID-19-Fällen und Kontrollen innerhalb von GSE147507 identifiziert. Wenn während des Konvertierungsprozesses einer Sonden-ID keine Gen-ID zugewiesen werden konnte, wurde die Sonden-ID nicht verwendet. Nach dem Zusammenführen vieler Sonden-IDs zu einer einzigen Gen-ID wurde die endgültige Berechnung als mittlerer Expressionswert bestimmt. Angepasste p-Werte von weniger als 0,05 und |logFC (Fold Change) |≥ 1 wurden als statistisch signifikant angesehen.DEGs-bezogene KEGG- und GO-AnalysenWir haben sowohl KEGG- als auch GO-Analysen durchgeführt, um die physiologischen Funktionen und funktionellen Zusammenhänge gemeinsamer COVID-19- und PH-DEGs besser zu verstehen. Angepasste p-Werte von < 0,05 wurden als signifikant erachtet. Die Top 10 DEGs wurden mit dem ClusterProfiler-Tool von R angezeigt.C-DEGs-bezogene Anreicherungs- und IdentifikationsanalysenMithilfe des VennDiagram-Tools (
jvenn.toulouse.inra.fr/app/example.html
) konnten wir C-DEGs (sowohl niedrig als auch hoch exprimierte) aus GSE147507 und GSE113439 visuell darstellen. Das Cluster-Profiler-Tool von R wurde verwendet, um die Ergebnisse der KEGG- und GO-Netzwerkstudien anzuzeigen. Die Signifikanz wurde wie zuvor definiert. Drei verschiedene maschinelle Lernalgorithmen – Support Vector Machine-Recursive Feature Elimination (SVM-RFE), Least Absolute Shrinkage and Selection Operator (LASSO) logistische Regression und Random Forests (RF)
72
,
73
,
74
,
75
,
76
,
77
– wurden verwendet, um potenzielle neue Biomarker für pädiatrische Sepsis zu ermitteln. Zusätzlich wurde die Random-Forest-Methode mit dem R-Paket „randomForest“ in R implementiert. Unter Verwendung des R-Pakets „glmnet“
78
wurde in dieser Studie eine LASSO-Logistik-Regressionsanalyse durchgeführt, wobei das minimale Lambda als das Beste erachtet wurde. Die partielle Wahrscheinlichkeitsabweichung lag unter 5 % und die Parameterauswahl für die Optimierung wurde mit einem Faktor von 10 überprüft. Die Gene, die Merkmale mit allen drei zuvor diskutierten Klassifizierungsschemata gemeinsam haben, wurden dann für die weitere Untersuchung ausgewählt.Nomogramm-Konstruktion und ROC-Kurve (Receiver Operating Characteristics)Die Expressionsniveaus der Kandidaten für Biomarker wurden zwischen der PH- und der Kontrollgruppe ausgewertet, sodass der Diagnosewert jedes Kandidaten mithilfe einer ROC-Kurve (Receiver Operating Characteristic) berechnet werden konnte. Der Diagnosewert wurde dann mithilfe eines 95%-Konfidenzintervalls und der Fläche unterhalb der ROC-Kurve (AUC) geschätzt. Um Verzerrungen zu reduzieren, wurde der GSE53408-Datensatz zur Validierung verwendet. Für die Nomogrammkonstruktion des rms R-Pakets wurden nur Kandidaten mit einer AUC > 0,7 in Test- und Validierungssätzen ausgewählt. AUC wurde verwendet, um die Diagnosegenauigkeit des Nomogramms zu überprüfen, und AUC sowie Entscheidungs- und Kalibrierungskurven wurden verwendet, um die Leistung des Nomogramms zu bewerten.Immuninvasion und GSEA-AnalyseAls nächstes wurde GSEA in R zur Auswertung der Hub-Gene verwendet, die nachgewiesen werden konnten. Die Bedeutung eines Phänotyps kann durch Auswertung des Genverteilungsmusters für dieses Merkmal anhand eines vordefinierten Gensatzes bestimmt werden. Um außerdem den Grad der Immuninfiltration in jeder Datensatzprobe zu messen, wurde der ssGSEA-Score zur Quantifizierung der Immunzellinvasion in COVID-19- und PH-Datensätzen und zur Feststellung der Immuninvasion innerhalb von GSE113439 und GSE147507 verwendet. Korrelationen zwischen 2 Kerngenen und 23 Immunzellen wurden mithilfe der Pearson-Korrelationsanalyse ermittelt, um potenzielle Korrelationen zwischen Immunzellen und Kerngenen aufzudecken
79
.TF-Gen- und TF-miRNA-modulatorische NetzwerkeDie Networkanalyst-Plattform (
www.networkanalyst.ca
) wurde zur Erstellung von TF-miRNA- und TF-Gen-Modulationsnetzwerken verwendet
80.
Anschließend validierten wir das TF-Gen- und TF-miRNA-Netzwerk mit einer FFL-Schleife. FFLtool ist verfügbar unter
100 ns Moleküldynamik-Simulationsanalyse des 1-Nitropyren-CCL20/SELE-Komplexes. ( A ) RMSD-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes, ( B ) Rg-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes, ( C ) RMSF-Kurve des 1-Nitropyren-CCL20-Komplexes, ( D ) RMSF-Kurve des 1-Nitropyren-SELE-Komplexes, ( E ) Wasserstoffbrückenänderungskurve des 1-Nitropyren-CCL20/SELE-Komplexes und ( F ) SASA-Kurve des 1-Nitropyren-CCL20/SELE-Komplexes.
DiskussionNach der ersten Beobachtung von Xie und Kollegen
12
, dass COVID-19 mit einem Anstieg von Herz-Kreislauf-Erkrankungen einhergeht, haben zahlreiche klinische Studien und Metaanalysen eine erhöhte Inzidenz von akuten Koronarsyndromen, Myokarditis, Perikarditis, Herzinsuffizienz und Arrhythmien bestätigt
13
,
14
,
15
,
16
,
17
,
18
,
19
,
20.
Zu den Folgen einer schweren COVID-19-Erkrankung gehören systemische Hypoxie, akutes Atemnotsyndrom, Hyperkoagulation, Sepsis, Entzündungen, metabolischer Stress und Zytokinstürme, die alle das Herz-Kreislauf-System belasten und schließlich zu einer Blutdruckentgleisung führen können
21
,
22
,
23
,
24
,
25
.Pulmonale Hypertonie als schwerwiegende kardiopulmonale Komplikation von COVID-19 erhöht die Wahrscheinlichkeit, dass eine Behandlung auf der Intensivstation, künstliche Beatmung, extrakorporale Membranoxygenierung (ECMO) oder sogar der Tod erforderlich werden. Daher könnte das frühzeitige Erkennen von hohem Pulmonalarteriendruck bei SARS-CoV-2-Patienten die Langzeitprognose der Patienten verbessern und die Hospitalisierungsrate sowie die Sterberate aufgrund solcher Komplikationen minimieren
7
,
8
,
9
,
26.
De Jong CMM
27
fand jedoch heraus, dass chronische thromboembolische pulmonale Hypertonie nach einer COVID-19-assoziierten Lungenembolie keine häufigere Langzeitkomplikation ist als nach einer nicht-COVID-19-assoziierten Lungenembolie. Unabhängig davon, ob dieses Phänomen auf große Unterschiede in der Pathogenese zurückzuführen ist, ist es besonders wichtig, die Pathogenese von COVID-19 in Kombination mit pulmonaler Hypertonie zu untersuchen.In dieser Studie wurde versucht, Moleküle zu untersuchen, die mit der Pathogenese von COVID in Kombination mit PH in Zusammenhang stehen, und zwar mithilfe verschiedener bioinformatischer Methoden. Anschließend wurde durch die Bewertung signifikanter Marker ein umfassendes Diagnosemuster erstellt. Die Expression jedes Gens wurde quantifiziert und bewertet, wobei höhere Werte mit einem größeren Vorhersagepotenzial verbunden waren. Diese Vorhersagewerte könnten zur Überwachung und Frühintervention bei COVID-19-Patienten, insbesondere bei Patienten mit PH, verwendet werden. Ein weiteres Ziel war die Identifizierung spezifischer Medikamente, die auf Schlüsselgene abzielen, die mit COVID-19 in Verbindung stehen und mit PH einhergehen, und so sowohl die Diagnose als auch die Behandlung dieser Erkrankung verbessern könnten.Ähnlich wie bei den genetischen Ursachen der pulmonal-arteriellen Hypertonie kann der Schweregrad von COVID-19 durch Variationen in denselben Genen beeinflusst werden. Viele der pathobiologischen Kennzeichen der pulmonal-arteriellen Hypertonie sind auch bei der durch COVID-19 induzierten pulmonalen Vaskulopathie vorhanden, darunter Endodermatitis, Vaskulitis-mediale Hypertrophie und Proliferation glatter Muskelzellen. Insbesondere eine endotheliale Dysfunktion ist ein häufiges Merkmal der bei COVID-19-Patienten beobachteten klinischen Manifestationen. Das Coronavirus SARS-CoV-2 gelangt in die Wirtszellen, indem es sein Spike-Glykoprotein an das Angiotensin-konvertierende Enzym 2 (ACE2), den Sialinsäurerezeptor, die transmembranöse Serinprotease 2 (TMPRSS2) und den Induktor der extrazellulären Matrix-Metalloproteinase (CD147) bindet; Catepsin B und L sind ebenfalls am Eindringen des Virus beteiligt. Alle diese Faktoren werden in Endothelzellen exprimiert
28
,
29
,
30
,
31
Überaktivierte Thrombozyten verursachen Zytokinstürme und Thrombosen. Studien haben gezeigt, dass Thrombozyten, die entzündungsfördernde Moleküle exprimieren und virale RNA tragen, besonders wahrscheinlich hochaktiv sind
32
.Unter den C-DEGs fanden wir beim Vergleich der COVID-19- und PH-Datensätze 47 stark exprimierte Gene und 15 schwach exprimierte Gene. Mithilfe der GO-Analyse stellten wir fest, dass C-DEGs mit den folgenden Begriffen angereichert waren: Entzündungsreaktion, immunologische Reaktion, Zelloberflächenrezeptor, zelluläre Reaktion auf Interferon-gamma, positive Regulierung von NF-kB und Reaktion auf Lipopolysaccharide. TNF-Signalweg, Infektion mit Herpes-simplex-Virus 1, virale Proteininteraktion mit Zytokin und Zytokinrezeptor, Malaria, Lipid und Atherosklerose, PPAR-Signalweg und PPAR-Signalweg waren alle unter den C-DEGs in der KEGG-Anreicherungsanalyse angereichert. Diese Ergebnisse legen stark nahe, dass eine Immunentzündung eine treibende Kraft bei der Entstehung und dem Fortschreiten von COVID-19 in Verbindung mit PH ist. Eine Infektion mit SARS-CoV-2 kann einen Zytokinsturm auslösen, der zu systemischer Entzündung und vaskulärer Endothelzellschädigung führt. Diese Veränderungen können Hyperkoagulabilität und intravaskuläre Thrombose verursachen und gleichzeitig den pulmonalen Gefäßwiderstand erhöhen. Diffuse Mikroangiopathie und Mikrothrombose, die durch eine umfassende Beeinträchtigung der vaskulären Endothelfunktion verursacht werden, können das Ungleichgewicht des Lungenventilations-/Blutflussverhältnisses weiter verschlimmern, und eine Erhöhung des pulmonalen Rechts-Links-Shunts kann die Hypoxie weiter verschlimmern und so die pulmonale Gefäßkontraktion und -umgestaltung fördern. Wenn der Körper von einem Virus angegriffen wird, kann dieser außerdem T-Zellen aktivieren und die Überexpression von IFN-induzierten Genen fördern, die ebenfalls zur Apoptose von Endothelzellen und damit zu PH führen können. Dies hat viele Gemeinsamkeiten mit den Ergebnissen der Studie von LuisG
33
.Um unter den 62 identifizierten DEGs weiter nach den Kerngenen zu suchen, die mit COVID-19 in Kombination mit PH in Verbindung stehen, verwendeten wir maschinelles Lernen, um potenzielle diagnostische Biomarker auszuwählen. Die LASSO-Regressionsanalyse identifizierte 20 Gene mit der geringsten binominalen Abweichung. Der Random-Forest-Ansatz identifizierte 10 Kandidaten, nachdem die DEGs nach Signifikanz eingestuft wurden, und die SVM-RFE-Methode identifizierte 8 Gene mit dem geringsten Fehler und der besten Genauigkeit nach 100-fachen Änderungen. Anhand der Schnittmenge der Ergebnisse der drei Algorithmen wurden SELE und CCL20 als Kerngene identifiziert. Es stellte sich heraus, dass diese beiden Schlüsselgene im Validierungssatz im Vergleich zueinander statistisch signifikante Unterschiede aufwiesen. Als Nächstes erstellten wir ein Vorhersagemodell, um noch fundiertere Vorhersagen zu COVID-19 in Kombination mit PH zu treffen, indem wir die Werte in der Tabelle auswerteten, wobei höhere Werte eine größere Wahrscheinlichkeit darstellten, nach COVID-19 an PH zu erkranken. Durch Zeichnen der ROC-, PR- und DCA-Kurven wurde festgestellt, dass das Vorhersagemodell zuverlässig war.Eines der identifizierten Kerngene war SELE. Die Aktivierung von Endothelzellen durch Zytokine führt zur Expression eines Zelloberflächen-Glykoproteins namens SELE, das die Anhaftung zirkulierender Monozyten und Lymphozyten an Endothelzellen erleichtert
34.
Der Plasmamarker für endotheliale Dysfunktion oder Verletzung ist lösliches E-Selectin (sE-Selectin), das von beschädigten oder dysfunktionalen Endothelzellen abgesondert wird
35
,
36.
Der Membrantransport von IFN-gamma-R2 zum Golgi-Apparat und die ordnungsgemäße IFN-gamma-R-Zusammensetzung erfordern beide die Beteiligung von E-Selectin. Die Aktivierung der BTK-Kinase wird durch die Interaktion eines E-selektiven Proteins ausgelöst, das wiederum einen funktionellen IFN-gamma-R bildet, der an einen anderen funktionellen IFN-gamma-R binden kann und so eine effiziente angeborene Reaktion von Makrophagen auf eine intrazelluläre bakterielle Infektion erzeugt
37
,
38
. Die Bedeutung von Endothelzellen bei der Verbreitung von SARS-CoV-2 wird zunehmend akzeptiert. Wahrscheinlich ist die Funktion von E-Selectin bei der Chemotaxis von Leukozyten während einer Entzündung der grundlegende Mechanismus, der hier am Werk ist. Die Expression von E-Selectin auf der Oberfläche nimmt zu, was den Eintritt von Leukozyten in das Gewebe und die Einleitung einer Entzündung zur Bekämpfung der Infektion erleichtern könnte
39
,
40.
DM Smadja et al. entdeckten, dass PH an zirkulierende Endothelzellen, lösliches E-Selectin und sVCAM gekoppelt ist, nicht jedoch an endotheliale Vorläuferzellen, CD34(+)CD133(+)-Zellen oder den vaskulären endothelialen Wachstumsfaktor (VEGF)
41.
Ähnlichkeiten zwischen diesen und den Ergebnissen unserer Studie legen nahe, dass SELE ein Schlüsselmolekül der pathogenen COVID-19-gekoppelten PH sein könnte.CCL20, auch bekannt als Makrophagen-Entzündungsprotein-3α oder durch Leberaktivierung reguliertes Chemokin, ist ein weiteres Gen von zentraler Bedeutung, das wir untersucht haben. CCL20 ist ein CC-Chemokin, das spezifisch mit CCR6 interagiert. Neben der Rekrutierung unreifer dendritischer Zellen, Effektor-/Gedächtnis-T-Zellen und B-Zellen spielt dieses Chemokin auch eine entzündungsfördernde Rolle bei der Aufrechterhaltung der Homöostase. Es spielt eine entscheidende Rolle bei der Aufrechterhaltung des regelmäßigen Verkehrs von Immunzellen und beim Auslösen einer T-Zell-abhängigen Entzündung. CCL20 reguliert das richtige Maß an Entzündung, indem es ein feines Gleichgewicht zwischen offensiver und defensiver Immunität aufrechterhält
42
,
43
,
44
,
45
. Es rekrutiert auch Th17-Zellen und regulatorische T-Zellen an Entzündungsherde, da CCR6 auf diesen Zelltypen vorhanden ist. Bei Patienten mit COVID-19 wurden sowohl in der bronchoalveolären Lavage (BAL)-Flüssigkeit als auch in Plasmaproben erhöhte CCL20-Werte festgestellt
46
. Pulmonalarterielle Hypertonie bei Personen mit SSc ist mit erhöhten CCL20-Serumspiegeln assoziiert
44
. Allerdings gibt es bisher nur wenig Forschung zu CCL20 bei COVID- und PH-Patienten.Die funktionelle Anreicherung betraf in erster Linie die adaptive Immunantwort, die durch Leukozyten und Lymphozyten vermittelte Immunantwort und die durch Zytokine wie IL-12 und TNF-a vermittelte entzündungsfördernde Reaktion, die die Proliferation von glatten Muskelzellen der Lungenarterie förderte und eine vaskuläre Umgestaltung herbeiführte, wie durch GSEA von SELE und CCL20 in den Datensätzen von COVID-19 und PH bestimmt wurde.Mithilfe einer ssGSEA-Analyse untersuchten wir die Beziehung zwischen SELE und CCL20 sowie 23 verschiedenen Arten von Immunzellen in den COVID-19- und PH-Datensätzen. Dabei stellten wir fest, dass aktivierte CD4-T-Zellen, aktivierte dendritische Zellen, natürliche Killer-T-Zellen, Neutrophile und plasmazytoide dendritische Zellen alle mit COVID-19 und PH in Verbindung standen. Die höchste Pearson-Korrelation wurde zwischen CCL20 und plasmazytoiden dendritischen Zellen und die höchste Verbindung zwischen SELE und aktivierten dendritischen Zellen festgestellt. Die immunologische Reaktion verschiedener Zellen kann durch SELE und CCL20 reguliert werden, was zu einer erhöhten Inzidenz von COVID-19-gekoppelter PH führt. Während einer akuten Coronavirus-Infektion werden Lymphozyten, einschließlich NK-Zellen, aktiviert und wandern in die Lunge, da sich dort entzündliche mononukleäre Makrophagen und Neutrophile ansammeln, die Zytokine und Chemokine freisetzen. Wenn die SELE- und CCL20-Werte sinken, nehmen Anzahl und Funktion der NK-Zellen ab. PH entwickelte sich infolge einer erhöhten Empfindlichkeit gegenüber COVID-19 und einer Veränderung der Lungenarterienwände durch die Zerstörung von NK-Zellen. Eine chronische Gewebeentzündung kann durch eine CD4+T-Zell-vermittelte zelluläre Immunität hervorgerufen werden, eine besondere zelluläre Immunantwort, die von CD4+T-Zellen gesteuert wird
47
,
48
,
49
,
50.
Es kommt zu einer Invasion von Lymphozyten (meistens T-Zellen) und mononukleären phagozytischen Zelllinien, was zu einer exsudativen Entzündung führt. Die Pathophysiologie der durch COVID-19 komplizierten PH wird auch durch SELE- und CCL20-vermittelte Neutrophilenproliferation beeinflusst. Durch die Sekretion von extrazellulären Fallen von Neutrophilen, die wiederum die Schädigung von Lungenarterien-Endothelzellen und die Proliferation von glatten Muskelzellen erhöhen, tragen Neutrophile dazu bei, die Entzündung sowohl aufrechtzuerhalten als auch zu verschlimmern.Laut einer Pearson-Analyse zeigten SELE, CCL20 und dendritische Zellen die größte Korrelation unter den fünf Zelltypen. Laut einer Forschung von Tomonari Sumi et al.
51
nahmen dendritische Zellen bei Personen mit COVID-19-Folgeerscheinungen dramatisch ab . Etwa 7 Monate nach der SARS-CoV-2-Infektion entdeckten Perez-Gomez A et al.
52
, dass die Zahl der dendritischen Zellen in vivo erheblich abnahm. Zwischen dieser Schlussfolgerung und unserer eigenen bestehen Parallelen. Bei Menschen mit COVID-19, die auch an PH leiden, ist die Zahl reifer myeloider DC und der damit verbundenen Funktionsstörungen zurückgegangen. Daher sind DC-Zellen nicht in der Lage, eine Immunreaktion einzuleiten, indem sie die Aktivierung und Vermehrung primärer T-Zellen fördern, um den Körper vor virusbedingten Schäden zu schützen.Neben der Diagnose ist auch die Identifizierung von Medikamenten, die auf die Pathogenese abzielen, eine wichtige Richtung für uns. Die derzeitige Behandlung von COVID-19 hängt in erster Linie von unterstützender Pflege sowie dem Einsatz antiviraler und immunmodulatorischer Medikamente ab. Angesichts der Verteilung der Bevölkerung mit Komorbiditäten, insbesondere der überwiegend mittleren und älteren Bevölkerungsgruppe, könnten Polypharmazeutika und Arzneimittelwechselwirkungen offensichtlich sein. Leider ist das potenzielle Risiko von Arzneimittelwechselwirkungen weitgehend unbekannt, da die meisten Studien zu COVID-19 keine Details zu Wechselwirkungen zwischen im Rahmen der COVID-19-Behandlung verwendeten Medikamenten und Komedikamenten zur Behandlung anderer Komorbiditäten bei diesen Patienten liefern
53
. Darüber hinaus kann die Verwendung einiger häufig verwendeter Medikamente bei COVID-Patienten zu einer erhöhten Anzahl unerwünschter Lungeneffekte führen
54
. Die derzeitige Richtung besteht darin, geeignete Medikamente zu finden, die eine bestimmte therapeutische Wirkung und relativ wenige Nebenwirkungen haben. Aufgrund ihrer Bedeutung für die Pathogenese von COVID-19 und PH haben wir in dieser Studie SELE und CCL20 als Ziele für die Arzneimittelvorhersage und das molekulare Docking ausgewählt. Die Bindungsenergie dieser beiden Moleküle mit FENRETINID, 1-NITROPYREN und AFLATOXIN B1 war die niedrigste aller acht erwarteten Medikamente, was nicht erwartet wurde. Derzeit gibt es einige Berichte, dass 1-NITROPYREN und AFLATOXIN B1 die Symptome von COVID-19 und PH lindern können
55
,
56
,
57
,
58
,
59
,
60
,
61
,
62.
Auch als Nitroverbindung fanden WenXia Feng et al. heraus, dass die Behandlung mit inhaliertem Stickoxid bei der Verringerung und Stabilisierung von PASP hilfreich war und auch das Risiko einer Rechtsherzinsuffizienz bei COVID-19 mit pulmonaler Hypertonie verringern könnte
63
. In dieser Studie stellten wir fest, dass wir durch die Durchführung von Medikamentenvorhersagen für die Schlüsselgene, die COVID mit PH kombiniert, sowohl in der Enricher-Datenbank als auch in der CTD-Datenbank vorhersagen konnten, dass 1-NITROPYREN ein potenzielles therapeutisches Medikament ist, und dass wir durch molekulares Docking und molekulare Dynamiksimulationen herausfanden, dass die Kombination von 1-NITROPYREN und den beiden Schlüsselgenen äußerst stabil war. Daher hat es großes Potenzial, als Medikament zur Behandlung dieser Komplikation eingesetzt zu werden. Aufgrund seiner positiven Auswirkungen auf die Glukosetoleranz, den Lipidspiegel und den Körperfettanteil wurde das synthetische Retinid-Derivat Fenretinid für eine Vielzahl von medizinischen Zwecken eingesetzt, darunter zur Krebsprävention und -behandlung, zur Verbesserung der Arteriosklerose und zur Linderung der nichtalkoholischen Fettlebererkrankung. Seine Fähigkeit, die Produktion von Entzündungsmediatoren zu reduzieren und die Makrophagenpolarisation zu verhindern, könnte hier der primäre Mechanismus sein
63
,
64
,
65.
Fenretinid hemmt die Freisetzung entzündungsfördernder Faktoren (IL-1β, MCP1, iNOS und TNF-α). Fenretinid könnte die NF-κB-Signalgebung hemmen, indem es die nukleare Translokation des Proteins durch Herunterregulierung der IKKβ- und IκBα-Phosphorylierung verringert
64.
Darüber hinaus ist die verzögerte Freisetzung von IFN-I bei einer SARS-CoV-Infektion als Mechanismus bekannt, der die antivirale Reaktion des Körpers verlangsamt. Die mit der Umgehung von IFN-I verbundenen viralen Mechanismen sind vielschichtig und umfassen die Sequestrierung und Abschirmung von RNA in Doppelmembranvesikeln, die Modifikation viraler mRNA-5-Cap-Strukturen und das spezifische Angreifen antiviraler zellulärer Signalwege. Bei SARS-CoV und MERS-CoV wirkt die IFN-I-Produktion nur in den frühen Stadien nach der Infektion schützend; zu späteren Zeitpunkten hingegen, wenn die Immunantwort verstärkt ist, werden IFN-I und inflammatorische Zytokine pathogen
66
,
67
,
68
,
69
. Sowohl beim Zika-Virus als auch beim Dengue-Virus hemmte Fenretinid das nichtstrukturelle Protein 5 (NS5), das zur Virulenz beitrug, indem es die Produktion von IFN-I verhinderte
70
. Daher ist es möglich, dass Fenretinid auch die Mechanismen beeinflusst, die die IFN-I-Umgehung bei Coronavirus-Infektionen regulieren. Es gibt jedoch noch wenige relevante Erkenntnisse und der genaue Wirkmechanismus ist unklar, sodass eingehendere pharmakologische Studien erforderlich sind.Unsere Literaturrecherche ergab, dass es an Forschung zum gemeinsamen Mechanismus von COVID-19 und PH mangelt, insbesondere an bioinformatischen Studien. Hier suchten wir nach C-DEGs, testeten Core-Gene mit einem maschinellen Algorithmus und erstellten ein Modell zur Vorhersage von COVID-19 in Kombination mit PH. Wir untersuchten, wie diese beiden wesentlichen Gene mit Krankheiten in Verbindung stehen, und machten Vorhersagen über die Transkriptionsfaktoren und miRNAs, die sie regulieren. Am Ende nutzten wir molekulares Docking und Targeting-Vorhersagen für zwei wichtige Gene, um zu bestimmen, welche Medikamente am wirksamsten wären.Unsere Forschung wies jedoch gewisse Lücken auf. Zunächst stellten wir durch molekulares Docking fest, dass Fenretinid, ein zielgerichtetes Medikament für SELE und CCL20, ein neues Ziel für die Behandlung der kombinierten PH mit COVID 19 sein könnte, obwohl der Wirkmechanismus noch weiter untersucht werden muss. Zur weiteren Verifizierung der aktuellen Ergebnisse ist eine externe Validierung erforderlich. Darüber hinaus ist eine In-vitro-Modellvalidierung erforderlich, um die Kerngenfunktionen zusätzlich zu erforschen.Materialen und MethodenDatenerfassungWir haben die gesamte GEO-Datenbank (
www.ncbi.nlm.nih.gov/geo/
) nach den Begriffen „COVID-19“ und „Pulmonale Hypertonie“
71
durchsucht . Alle sequenzierten Daten stammten von Menschen, und alle gescreenten Datensätze umfassten sowohl Kontrollpersonen als auch erkrankte Personen. Die Datensätze GSE113439, GSE147507 und GSE53408 haben einen rigorosen Screeningprozess durchlaufen und wurden schließlich ausgewählt. GSE147507 enthielt insgesamt 23 Patienten mit COVID-19 und Proben von 55 gesunden Freiwilligen, während GSE113439 15 Patienten mit PH und 11 normale Kontrollpersonen umfasste. Außerdem gab es im Datensatz GSE53408 11 gesunde Personen und 12 mit sehr schwerer PH, sowie 9 gesunde Personen und 26 mit COVID-19 im Datensatz GSE196822. Eine Übersicht dieser Datensätze finden Sie in Tabelle
S8
. Das genomische Profil der DEGs wurde log2-transformiert und Gensymbole wurden mithilfe von Annotationsdokumentationen aus relevanten Datensätzen mit Sonden abgeglichen. Schließlich wurde eine Genmatrix zur weiteren Analyse extrahiert, wobei Gensymbole in Spalten und Probennamen in Zeilen dargestellt wurden.DEG-ScreeningDEGs wurden zwischen PH und Kontrollen im GSE113439-Datensatz mithilfe der Pakete Limma und GEOquery im R-Programm sowie zwischen COVID-19-Fällen und Kontrollen innerhalb von GSE147507 identifiziert. Wenn während des Konvertierungsprozesses einer Sonden-ID keine Gen-ID zugewiesen werden konnte, wurde die Sonden-ID nicht verwendet. Nach dem Zusammenführen vieler Sonden-IDs zu einer einzigen Gen-ID wurde die endgültige Berechnung als mittlerer Expressionswert bestimmt. Angepasste p-Werte von weniger als 0,05 und |logFC (Fold Change) |≥ 1 wurden als statistisch signifikant angesehen.DEGs-bezogene KEGG- und GO-AnalysenWir haben sowohl KEGG- als auch GO-Analysen durchgeführt, um die physiologischen Funktionen und funktionellen Zusammenhänge gemeinsamer COVID-19- und PH-DEGs besser zu verstehen. Angepasste p-Werte von < 0,05 wurden als signifikant erachtet. Die Top 10 DEGs wurden mit dem ClusterProfiler-Tool von R angezeigt.C-DEGs-bezogene Anreicherungs- und IdentifikationsanalysenMithilfe des VennDiagram-Tools (
jvenn.toulouse.inra.fr/app/example.html
) konnten wir C-DEGs (sowohl niedrig als auch hoch exprimierte) aus GSE147507 und GSE113439 visuell darstellen. Das Cluster-Profiler-Tool von R wurde verwendet, um die Ergebnisse der KEGG- und GO-Netzwerkstudien anzuzeigen. Die Signifikanz wurde wie zuvor definiert. Drei verschiedene maschinelle Lernalgorithmen – Support Vector Machine-Recursive Feature Elimination (SVM-RFE), Least Absolute Shrinkage and Selection Operator (LASSO) logistische Regression und Random Forests (RF)
72
,
73
,
74
,
75
,
76
,
77
– wurden verwendet, um potenzielle neue Biomarker für pädiatrische Sepsis zu ermitteln. Zusätzlich wurde die Random-Forest-Methode mit dem R-Paket „randomForest“ in R implementiert. Unter Verwendung des R-Pakets „glmnet“
78
wurde in dieser Studie eine LASSO-Logistik-Regressionsanalyse durchgeführt, wobei das minimale Lambda als das Beste erachtet wurde. Die partielle Wahrscheinlichkeitsabweichung lag unter 5 % und die Parameterauswahl für die Optimierung wurde mit einem Faktor von 10 überprüft. Die Gene, die Merkmale mit allen drei zuvor diskutierten Klassifizierungsschemata gemeinsam haben, wurden dann für die weitere Untersuchung ausgewählt.Nomogramm-Konstruktion und ROC-Kurve (Receiver Operating Characteristics)Die Expressionsniveaus der Kandidaten für Biomarker wurden zwischen der PH- und der Kontrollgruppe ausgewertet, sodass der Diagnosewert jedes Kandidaten mithilfe einer ROC-Kurve (Receiver Operating Characteristic) berechnet werden konnte. Der Diagnosewert wurde dann mithilfe eines 95%-Konfidenzintervalls und der Fläche unterhalb der ROC-Kurve (AUC) geschätzt. Um Verzerrungen zu reduzieren, wurde der GSE53408-Datensatz zur Validierung verwendet. Für die Nomogrammkonstruktion des rms R-Pakets wurden nur Kandidaten mit einer AUC > 0,7 in Test- und Validierungssätzen ausgewählt. AUC wurde verwendet, um die Diagnosegenauigkeit des Nomogramms zu überprüfen, und AUC sowie Entscheidungs- und Kalibrierungskurven wurden verwendet, um die Leistung des Nomogramms zu bewerten.Immuninvasion und GSEA-AnalyseAls nächstes wurde GSEA in R zur Auswertung der Hub-Gene verwendet, die nachgewiesen werden konnten. Die Bedeutung eines Phänotyps kann durch Auswertung des Genverteilungsmusters für dieses Merkmal anhand eines vordefinierten Gensatzes bestimmt werden. Um außerdem den Grad der Immuninfiltration in jeder Datensatzprobe zu messen, wurde der ssGSEA-Score zur Quantifizierung der Immunzellinvasion in COVID-19- und PH-Datensätzen und zur Feststellung der Immuninvasion innerhalb von GSE113439 und GSE147507 verwendet. Korrelationen zwischen 2 Kerngenen und 23 Immunzellen wurden mithilfe der Pearson-Korrelationsanalyse ermittelt, um potenzielle Korrelationen zwischen Immunzellen und Kerngenen aufzudecken
79
.TF-Gen- und TF-miRNA-modulatorische NetzwerkeDie Networkanalyst-Plattform (
www.networkanalyst.ca
) wurde zur Erstellung von TF-miRNA- und TF-Gen-Modulationsnetzwerken verwendet
80.
Anschließend validierten wir das TF-Gen- und TF-miRNA-Netzwerk mit einer FFL-Schleife. FFLtool ist verfügbar unter
- Qingbin Hou ,
- Jinping Jiang ,
- Kun Na ,
- Xiaolin Zhang ,
- Daniel Liu ,
- Quanmin Jing ,
- Chenghui Yan &
- Han-Yan-Ling
- 849 Zugriffe
- 1 Altmetric
- MetrikenEinzelheiten
zwischen COVID-19-Patienten und Kontrollen. Wie in Abb. 2B gezeigt werden kann, konnten im GSE113439-Datensatz 461 stark exprimierte und 81 kaum exprimierte Gene beobachtet werden, die PH-Patienten von gesunden Kontrollen unterscheiden. Mithilfe des KEGG fanden wir heraus, dass der PH-Datensatz angereichert war mit dem Prozess der Regulierung des Aktin-Zytoskeletts (Padj = 0,031), der Ribosomenbiogenese in Eukaryoten (Padj = 0,0073), des RNA-Abbaus (Padj = 0,0072), des nukleozytoplasmatischen Transports (Padj = 0,022), der Kontraktion der Gefäßglattmuskulatur (Padj = 0,038) und der Motorproteine (Padj = 0,022). Es lag eine hypertrophe Kardiomyopathie (Padj = 0,014), eine Spondyloarthritis (Padj = 0,017) und eine rechtsventrikuläre Kardiomyopathie aufgrund von Arrhythmie (Padj = 0,022) vor (Abb.
2
C). Der COVID-19-Datensatz hingegen wurde angereichert mit einer Infektion mit Herpes-simplex-Virus 1 (Padj = 0,000053), einer Interaktion zwischen Zytokinen und ihren Rezeptoren (Padj = 0,00000002), einer Interaktion zwischen neuroaktiven Liganden und Rezeptoren (Padj = 0,00086), einem PI3K-Akt-Signalweg (Padj = 0,016), Lipid- und atherosklerotischen Erkrankungen (Padj = 0,000044), Tuberkulose (Padj = 0,00000083), einem Chemokin-Signalweg (Padj = 0,0000051), einem NOD-ähnlichen Rezeptor-Signalweg (Padj = 0,000013) und COVID-19 (Padj = 0,025) (Abb.
2
D). Darüber hinaus wurden bei der Analyse von Genen unter Verwendung von GO-Begriffen folgende Anreicherungen von DEGs im COVID-Datensatz als Reaktion auf eine Entzündungsreaktion (Padj = 7,20 × 10–26 ) , eine Immunreaktion (Padj = 8,40 × 10–16 ) , eine Chemotaxis von Neutrophilen (Padj = 4,00 × 10–14 ) , eine Zell-Zell-Signalisierung (Padj = 8,30 × 10–12 ) und eine zelluläre Reaktion auf Lipopolysaccharide (Padj = 1,30 × 10–11 ) beobachtet (Abb.
2
E). Die Förderung von RNA-Polymerase II (Padj = 2,30 × 10–03 ) , Zellteilung (Padj = 1,30 × 10–05 ) , Proteinphosphorylierung (Padj = 8,50 × 10–03 ) , Zelladhäsion (Padj = 0,015), zellulärer Reaktion auf DNA-Schäden (Padj = 3,30 × 10–06 ) , DNA-Reparatur (Padj = 4,40 × 10–06 ) , Entzündungsreaktion (Padj = 0,005) und negativer Regulation des apoptotischen Prozesses (Padj = 0,038) wurden im PH-Datensatz angereichert, wie in Abb.
2
F gezeigt.Abbildung 2
waren.Abbildung 3
. Der Random-Forest-Ansatz wurde verwendet, um 10 Kandidaten zu identifizieren, nachdem die DEGs nach Gensignifikanzbewertung eingestuft wurden (Abb.
4
C,D). Für die PH-Progression bei COVID19 identifizierte die SVM-RFE-Methode 8 Gene mit dem geringsten Fehler und der besten Genauigkeit nach 100-facher Verwendung (Abb.
4
E,F). Schließlich wurde ein Venn-Diagramm verwendet, um zu zeigen, wie sich SELE und CCL20 (Abb.
4
G) als DEGs erwiesen, wenn beide Methoden zusammen verwendet wurden.Abbildung 4
. Nach mehreren Auswahliterationen wurden SELE und CCL20 ausgewählt, um ein Nomogramm zu bilden (Abb.
5
C). Der Expressionsgrad jedes Gens wurde im Nomogramm mit einem numerischen Wert versehen. Schließlich wurde der Gesamtwert angewendet, um die Inzidenz des PH-Fortschritts bei COVID-19-Patienten vorherzusagen (Abb.
5 D). Das Nomogramm zeigte mit einem AUC von 0,826 (95 % KI 0,637–1.000; Abb.
5
D) außergewöhnlich gute Ergebnisse bei der Vorhersage der PH-Entwicklung bei COVID19. Darüber hinaus wurden auch ein Präzisions-Recall (PR) und eine Entscheidungskurvenanalyse (DCA) für das Nomogramm durchgeführt. Diese zeigten, dass das Nomogrammmodell für die Diagnose von COVID-19 mit PH von Nutzen sein kann (Abb.
5
E,F).Abbildung 5
und GSE147507 (Abb.
6
C,D) stark mit aktivierten Immunreaktionen wie der adaptiven Immunreaktion um Blutgefäße herum verbunden waren. Der Anstieg des proinflammatorischen Faktors erhöhte die Expression von Zytokinen und Chemokinen, was zu einer übermäßigen Kontraktion und Proliferation von Blutgefäßzellen beitrug.Abbildung 6
.Abbildung 7
In der Kohorte GSE113439 korrelierten CCL20 und SELE positiv mit vielen Zelltypen. Einschließlich: Typ-1-T-Helferzelle, regulatorische T-Zelle, plasmazytoide dendritische Zelle, Neutrophil, natürliche Killer-T-Zelle, Mastzelle, zentrale Gedächtnis-CD4-T-Zelle, aktivierte dendritische Zelle und aktivierte CD4-T-Zelle. Rot: positive Korrelation; Blau: negative Korrelation. ( C ) Laut einer Korrelationsstudie von Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Verknüpfungsgrad mit SELE aufwiesen.
Als wir die ssGSEA-Ergebnisse aus den oben genannten Datensätzen kombinierten, stellten wir fest, dass COVID-19 und PH mit der Aktivierung von CD4-T-Zellen, aktivierten dendritischen Zellen, natürlichen Killer-T-Zellen, Neutrophilen und plasmazytoiden dendritischen Zellen in Zusammenhang standen. Laut einer Korrelationsstudie nach Pearson wiesen plasmazytoide dendritische Zellen den höchsten Assoziationsgrad mit CCL20 auf, während aktivierte dendritische Zellen den höchsten Kopplungsgrad mit SELE aufwiesen (Abb.
7
C).Komodulatorische Achse TF-miRNA und TF-Gen-AssoziationenAnschließend wurde der Netzwerkanalyst verwendet (Abb.
8
A). Das erstellte Netzwerk enthielt insgesamt 19 Knoten und 18 Kanten. Es gab eine starke Korrelation zwischen Hub-Genen und TFs, und die TFs beeinflussten mehr als ein Hub-Gen im Netzwerk. Um die Verbindung zwischen miRNAs + TFs und Hub-Genen zu bewerten, erstellten wir die TF-miRNA-Komodulatorachse. Unser Netzwerk enthielt 39 Knoten, 38 Kanten und 25 miRNA-basierte Interaktionen, an denen Hub-Gene beteiligt waren, wodurch die Gesamtexpression der Hub-Gene moduliert werden konnte. In unserer Forschung wurden SELE und CCL20 als potenzielle diagnostische Biomarker für COVID-19 in Verbindung mit pulmonaler Hypertonie (PH) identifiziert. Um die Rolle dieser Gene bei der Krankheit weiter zu validieren, schlagen wir die Verwendung des FFLtools zur Analyse von Transkriptionsfaktor-Gen- und Transkriptionsfaktor-miRNA-Netzwerken vor. FFLtool ist ein Webserver, der für die Analyse von Feedforward-Loops (FFLs) entwickelt wurde und tiefe Einblicke in die Wechselwirkungen zwischen Transkriptionsfaktoren, miRNAs und Genen bietet. Durch die Einbeziehung unserer Schlüsselgene, Transkriptionsfaktoren und miRNAs in diese Analyse. Als Ergebnis erschien die FFL, die TF CCL20, miRNA miR-1256 und das Zielgen PPARG enthält, ganz oben (Abb.
8
B, ergänzende Abb.
1
). Diese FFL unter CCL20, miR-1256 und PPARG könnte ein neuartiges regulatorisches Modul bei COVID-19 sein, das durch pulmonale Hypertonie kompliziert wird.Abbildung 8
. Die niedrigen Rg-Werte weisen darauf hin, dass das Protein während des Simulationsprozesses von 100 ns kompakt blieb. Im 1-Nitropyren-CCL20-Komplex schwankte der RMSF der C-Kette des CCL20-Proteins erheblich; dies war besonders im Bereich der Aminosäuren 27–36 zu beobachten, der, wie aus der Proteinstruktur ersichtlich, in der Schleifenregion der C-Kette liegt, was zu einer größeren Flexibilität während des 100-ns-Prozesses führt. Die RMSF-Werte der α-Helix und der β-Faltung in der C-Kette blieben während des Simulationsprozesses relativ niedrig, während die RMSF-Werte der A- und B-Ketten sich im Wesentlichen gleichzeitig änderten, was darauf hindeutet, dass das kleine 1-Nitropyren-Molekül wenig Einfluss auf die Gesamtflexibilität des CCL20-Proteins hatte. Im 1-Nitropyren-SELE-Komplex wurden große RMSF-Werte im Bereich der 124–140 Aminosäuren beobachtet, der laut molekularem Docking die Bindungstasche des kleinen Moleküls umgibt. Daher spekulieren wir, dass die Zugabe von 1-Nitropyren die Flexibilität der Aminosäuren an der Bindungsstelle erhöhte. Die Ergebnisse zeigen einen großen RMSF-Wert (Abb.
10
C,D). Im 100-ns-Simulationsprozess blieb die Anzahl der Wasserstoffbrücken im 1-Nitropyren-CCL20/SELE-Komplex bei 2, während die maximale Anzahl von Wasserstoffbrücken im 1-Nitropyren-CCL20-Komplex 3 betrug. Der 1-Nitropyren-SELE-Komplex könnte bis zu fünf Wasserstoffbrücken aufweisen. Diese beträchtliche Anzahl von Wasserstoffbrücken trägt dazu bei, die Stabilität des Komplexes aufrechtzuerhalten (Abb.
10
E). Die SASA-Kurven der 1-Nitropyren-CCL20- und 1-Nitropyren-SELE-Komplexe blieben während des gesamten Prozesses stabil und die mittleren SASA-Werte waren wie folgt: Die niedrigen SASA-Werte von 128 nm 2 und 92 nm 2 (Abb.
10
F).Abbildung 10
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.