
- Beiträge: 1755
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Zusammenhang zwischen Entzündung und Verhärtung der Lungenarterien
Zusammenhang zwischen Entzündung und Verhärtung der Lungenarterien
13 Aug 2024 12:52 #2174
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Zusammenhang zwischen Entzündung und Verhärtung der Lungenarterien wurde erstellt von danny
www.frontiersin.org/journals/immunology/...959209/fullPulmonale Hypertonie (PH) ist eine fortschreitende Erkrankung, die mehrere Ursachen haben kann und letztendlich zu Rechtsherzversagen als Hauptursache für Morbidität und Mortalität führt. Bei Patienten sind unterschiedliche Entzündungsreaktionen ein hervorstechendes Merkmal bei verschiedenen Arten von PH, und es wurde gezeigt, dass verschiedene immunmodulatorische Interventionen die Entwicklung und den Verlauf der Krankheit in Tiermodellen modulieren. Insbesondere umfasst die mit PH verbundene Entzündung die Infiltration sowohl angeborener als auch adaptiver Immunzellen in die Gefäßwand des Lungengefäßsystems – insbesondere bei Lungengefäßläsionen – sowie erhöhte Zytokin- und Chemokinspiegel im zirkulierenden Blut und im perivaskulären Gewebe der Lungenarterien (PAs). Frühere Studien legen nahe, dass veränderte hämodynamische Kräfte eine endotheliale Lungenfunktionsstörung und damit die Anhaftung von Immunzellen und die Freisetzung von Entzündungsmediatoren verursachen, während die daraus resultierende perivaskuläre Entzündung wiederum die Gefäßumgestaltung und das Fortschreiten der PH fördert. So kann sich ein Teufelskreis aus Endothelaktivierung, Entzündung und Gefäßumbau entwickeln, der den Krankheitsverlauf vorantreibt. Die PA-Versteifung ist ein aufstrebender Forschungsbereich bei PH und relevant für die PH-Diagnostik, Prognose und als therapeutisches Ziel. Hinsichtlich ihres prognostischen Werts konkurriert die PA-Versteifung mit der gut etablierten Messung des pulmonalen Gefäßwiderstands als Prädiktor des Krankheitsausgangs. Vaskulärer Umbau der arteriellen extrazellulären Matrix (ECM) sowie Gefäßverkalkung, Versteifung glatter Muskelzellen, Verdickung der Gefäßwände und Gewebefibrose tragen zur PA-Versteifung bei. Während Assoziationen zwischen Entzündungen und Gefäßversteifung bei systemischen Gefäßerkrankungen wie Arteriosklerose oder den vaskulären Manifestationen der systemischen Sklerose gut belegt sind, wurde ein ähnlicher Zusammenhang zwischen entzündlichen Prozessen und PA-Versteifung im Zusammenhang mit PH bislang nicht untersucht. In dieser Übersicht diskutieren wir mögliche Zusammenhänge zwischen Entzündungen und PA-Versteifung mit besonderem Schwerpunkt auf Gefäßverkalkung und ECM-Umbau bei PH. EinführungPulmonale Hypertonie (PH) umfasst eine Gruppe von Erkrankungen, bei denen der mittlere pulmonalarterielle Druck (mPAP) gemäß den aktuellen Leitlinien in Ruhe 25 mmHg überschreitet (
1
). Das vor kurzem stattgefundene 6. Weltsymposium zur PH empfahl, diesen Grenzwert weiter auf 20 mmHg abzusenken (
2
). Die Weltgesundheitsorganisation (WHO) teilt PH anhand erkennbarer Ursachen und Risikofaktoren in fünf Gruppen ein (
3
). Obwohl die Behandlung der pulmonal arteriellen Hypertonie (PAH) (WHO-Gruppe 1) das Stadium der zielgerichteten Therapie erreicht hat, beträgt die 5-Jahres-Überlebensrate von Patienten mit PAH immer noch lediglich etwa 50 % (
4
). Dies ist vermutlich auf die multifaktoriellen pathophysiologischen Mechanismen der PAH zurückzuführen, die sich einer gezielten Behandlung durch ein einzelnes pharmakologisches Medikament entziehen, insbesondere im fortgeschrittenen Krankheitsstadium (
5
). Daher bleibt das ultimative Ziel für eine verbesserte Versorgung von PH-Patienten die Identifizierung und therapeutische Ausrichtung gemeinsamer vorgelagerter Mechanismen, die mehrere nachgelagerte zelluläre und molekulare Prozesse auslösen, die die Umgestaltung der Lungengefäße in verschiedenen PH-Gruppen steuern.In letzter Zeit hat die pulmonale perivaskuläre Entzündung als frühes gemeinsames Kennzeichen verschiedener PH-Gruppen allmählich an Aufmerksamkeit gewonnen. Im Frühstadium der Erkrankung weisen PAH-Patienten und entsprechende Tiermodelle nicht nur eine Ansammlung von Immunzellen wie Makrophagen (
6
,
7
) und Mastzellen (
8
) in ihren Lungen (
9
) auf, sondern auch erhöhte Konzentrationen von Entzündungsmediatoren in ihrem Lungenkreislauf (
10
,
11
) (
Abbildung 1
). Bei den meisten Formen von PH ist diese Entzündungsreaktion vorwiegend auf die pulmonale Adventitia beschränkt (
7
). Tatsächlich neigen Veränderungen in der Adventitia, die aus einem komplexen Gemisch heterogener Zellen besteht, dazu, denen in anderen Gefäßkompartimenten vorauszugehen und sind für die Gefäßumgestaltung erforderlich (
12
). Bei PAH wurde diese räumliche Prädilektion mit der Tatsache in Verbindung gebracht, dass Fibroblasten in der pulmonalen Adventitia einen entzündungsfördernden Phänotyp mit einer erhöhten Expression von Entzündungsmediatoren aufweisen, die die Rekrutierung angeborener Immunzellen vorantreiben (
7
,
13
,
14
). Die daraus resultierende perivaskuläre Entzündung wird heute als entscheidender Pathomechanismus angesehen, der eine Umgestaltung von außen nach innen orchestriert, nicht nur bei PH, die mit Erkrankungen des Immunsystems einhergeht, wie z. B. der pulmonal-arteriellen Hypertonie in Verbindung mit Bindegewebserkrankungen (CTD-PAH) (
15
), sondern auch bei anderen Formen von PAH (
11
,
16
) sowie bei PH aufgrund einer Linksherzerkrankung (PH-LHD) (
17
). Parallel dazu setzt die Adventitia eine Vielzahl von Faktoren frei, die Differenzierung, Proliferation, Apoptose, Migration und Kollagensynthese durch andere Zellen in der Gefäßwand regulieren, während sich Adventitiafibroblasten in Myofibroblasten verwandeln und durch die mediale Schicht in die Intima migrieren können (
12
). Daher wurde angenommen, dass entzündliche Prozesse den Gefäß- und Immunzellstoffwechsel verändern und letztlich den Umbau der Pulmonalarterie (PA) verstärken und die PH verschlimmern (
Abbildung 1
). ABILDUNG 1
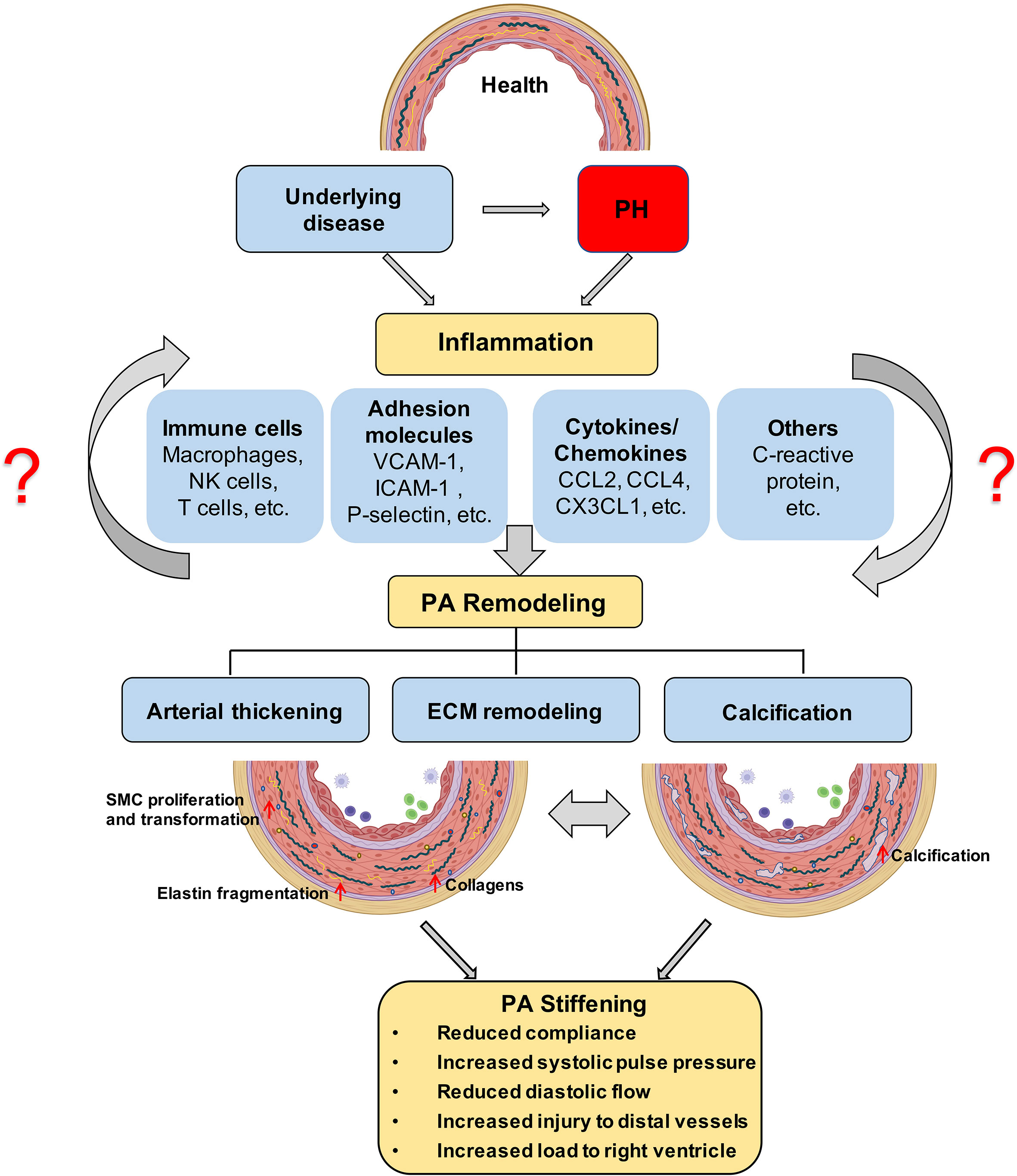 Abbildung 1 Vorgeschlagene Rolle der Entzündung bei der PA-Versteifung; Die Entwicklung von PH ist mit einer Entzündungsreaktion im Lungengefäßsystem verbunden, die durch Infiltration von Immunzellen und die Sekretion von Immunfaktoren gekennzeichnet ist. Eine ECM-Versteifung, insbesondere eine proximale großflächige Lungengefäßsklerose, tritt in den frühen Stadien von PH auf und hat einen prognostischen Wert für den Patientenerfolg und die spätere Verkalkung und wird durch Entzündungen verursacht. Eine anhaltende Angiosklerose fördert wiederum eine Entzündungsreaktion, die die Verkalkung und Verdickung der Lungengefäße verschlimmert. PH, pulmonale Hypertonie; ECM, extrazelluläre Matrix; PA, Lungenarterie. Gleichzeitig hat sich im letzten Jahrzehnt die PA-Versteifung als frühes Kennzeichen, Pathomechanismus und Prädiktor von Morbidität und Mortalität bei PH herausgestellt (
18
–
20
). Die Gefäßversteifung, definiert als erhöhter Widerstand der Arterienwand gegen Verformung während des Bluteinflusses, ist eine Folge pathologischer Gefäßumgestaltung, die sowohl in großen proximalen Arterien als auch in kleinen distalen Arterien und Arteriolen auftreten kann. Die mechanischen Folgen dieser Strukturveränderungen sind eine verringerte Compliance in proximalen PAs und ein erhöhter Widerstand gegen den Blutfluss (pulmonaler Gefäßwiderstand, PVR) in distalen PAs (
21
). Die PA-Compliance (PAC) ist wichtig, um den pulsierenden Blutfluss, der über den Windkessel-Effekt in die großen Conduit-Arterien gelangt, in eine nahezu laminare Strömung auf Höhe des distalen pulmonalen Gefäßbaums umzuwandeln. So reduziert PAC die Nachlast des rechten Ventrikels (RV) und hält die Lungendurchblutung über den Herzzyklus hinweg nahezu konstante aufrecht. In Übereinstimmung mit dem Einfluss von PAC auf die RV-Funktion hat die invasive oder nicht-invasive Bewertung von PAC (oder Kapazität) ergeben, dass die PA-Versteifung bei PAH-Patienten ein sensitiver Prädiktor für pathologische RV-Umgestaltung und Mortalität ist (
21
-
24
). Des Weiteren wurde vorgeschlagen, dass eine Versteifung proximaler PAs durch eine Erhöhung der Pulswellengeschwindigkeit und der vom Blut ausgeübten Scherspannung Verletzungen und Umgestaltungen in distalen Gefäßen fördert und so die Pathologie der PH in einer positiven Feedforward-Schleife vorantreibt (
25
). Eine derartige Interdependenz zwischen proximalen und distalen PA-Regionen würde voraussagen, dass pathologische Umgestaltungen in großen und kleinen Gefäßen parallel erfolgen sollten. Tatsächlich wurde in einer Arbeit von Stuart R. Reuben erstmals eine hyperbolische Beziehung zwischen PAC und PVR festgestellt (
26
). Das Produkt aus PAC x PVR ergibt die Resistance-Compliance-Zeit (RC-Zeit), die bei PH-Patienten der WHO-Klasse I (PAH), III (PH aufgrund einer chronischen Lungenerkrankung), IV (chronisch thromboembolische PH) oder V (PH mit unklaren multifaktoriellen Mechanismen) als nahezu konstant und unabhängig von der medikamentösen Therapie angesehen wird (
27
). Interessanterweise ist jedoch bei Patienten mit PH der WHO-Klasse II (PH aufgrund einer Linksherzerkrankung) die RC-Zeit reduziert, d. h. für jeden gegebenen PVR ist die entsprechende PAC niedriger als bei PH-Patienten mit anderen Ursachen. Bemerkenswerterweise ist diese Reduzierung der RC-Zeit auch mit einer Erhöhung der RV-Nachlast verbunden (
27
). Dieser interessante Befund könnte auf andere Pathomechanismen und/oder eine stärkere Versteifung der proximalen PAs bei PH-Patienten mit zugrunde liegender Linksherzerkrankung im Vergleich zu anderen Formen der PH hinweisen; diese Annahme muss jedoch noch streng getestet und mechanistisch untersucht werden.Umgekehrt kann eine mechanische Kommunikation zwischen proximalen PAs und dem distalen Lungengefäßsystem auch die Wiederherstellung der pulmonalvaskulären Homöostase fördern. Hinweise auf einen solchen umgekehrten Umbauprozess stammen aus einigen klinischen Studien bei Patienten mit angeborenen Herzfehlern und PH aufgrund von intrakardialen Links-Rechts-Shunts, die eine Überperfusion der Lunge verursachen. Bei diesen Patienten konnte eine chirurgische Bandierung des PA – durchgeführt mit der Absicht, den proximalen PA vor übermäßigem Druck und Fluss zu schützen – die PH erfolgreich verbessern und in einigen Fällen den vaskulären Umbau in den distalen Arterien umkehren (
28
,
29
).Eine wachsende Zahl von Studien über Techniken zur Abschätzung der Steifigkeit proximaler PAs in vivo zeigt, dass die Abschätzung der PA-Steifigkeit als Prognoseinstrument bei PH vielversprechend ist. Am häufigsten wird die PA-Steifigkeit durch Berechnung der pulmonalarteriellen Kapazität als Verhältnis des Schlagvolumens zum pulmonalen Pulsdruck abgeschätzt, die entweder durch Herzkatheterisierung oder nicht-invasiv durch Echokardiographie ermittelt wird (
20
,
30
–
37
), oder durch Berechnung eines Steifigkeitsindex als Änderung des PA-Drucks (wiederum ermittelt durch Rechtsherzkatheterisierung) geteilt durch die entsprechende Änderung des PA-Durchmessers (bestimmt durch Echtzeit-Bildgebungsverfahren wie kardiale Magnetresonanztomographie) (
18
,
38
).Die Arterienversteifung bei Herz-Kreislauf-Erkrankungen wird hauptsächlich auf eine Umgestaltung der extrazellulären Matrix (ECM) und/oder Verkalkung in der Arterienwand zurückgeführt (
39
–
42
) (
Abbildung 1
). PAH ist insbesondere durch eine Umgestaltung der ECM und eine Verdickung aller drei Schichten der PA-Wand gekennzeichnet (
43
), was letzten Endes die arterielle Compliance reduziert. Die PAs von PAH-Patienten weisen eine erhöhte Ablagerung von interstitiellem Kollagen auf, darunter Kollagen I, Kollagen XIV und Basalmembran-spezifische Kollagene, insbesondere Kollagen IV (
43
–
45
). Darüber hinaus wurde bei PAH-Patienten eine erhöhte Expression anderer ECM-Proteine wie Elastin und Fibronektin oder des matrizellulären ECM-Proteins Tenascin-C durch dedifferenzierte Adventitiafibroblasten beobachtet (
46
). Eine erhöhte Produktion und Ablagerung von ECM-Bestandteilen in PAs wird als adaptive Reaktion auf die erhöhte Verdauung von ECM der Media- und Basalmembran (BM) durch Matrix-Metalloproteinasen (MMPs) angesehen, die bei Patienten mit PAH (
47
) und IPAH (
45
) erhöht sind. Die erhöhte Expression von Kollagen durch Endothelzellen (ECs), glatte Muskelzellen (SMCs) und Adventitiafibroblasten ist mit einer erhöhten Kollagenvernetzung durch Lysyloxidasen (LOXs) verbunden (
48
). Außerdem induzieren proteolytische Enzyme auch den Abbau elastischer Fasern, deren Wiederaufbau trotz erhöhter Elastin-Genexpression aufgrund der komplexen 3D-Mehrkomponentenstruktur dieser Fasern schwierig ist (
49
–
53
). Die PA-Versteifung ergibt sich also als fortschreitendes Ungleichgewicht von Kollagen- gegenüber Elastinfaserkomponenten in der PA-Wand.Eine Gefäßversteifung wird auch auf Gefäßverkalkung (
40
) zurückgeführt, eine pathologische Ablagerung von festen Mineralien in der Intima oder Media der Arterienwände (
54
) (
Abbildung 1
). Wichtig ist, dass die Verkalkung von Lungengefäßen mit einer Transdifferenzierung von SMCs in osteogene Linien in Zusammenhang gebracht wurde, die durch die Aktivität des pro-osteogenen Transkriptionsfaktors RUNX2 (Runt-related transcription factor 2) (
55
) verursacht wird. Eine erhöhte nukleäre Expression von RUNX2 in PA-SMCs aktiviert nicht nur die Expression von verkalkungsbezogenen Biomineralisierungsgenen (
56
), sondern fördert auch die Zellproliferation und Apoptoseresistenz durch Aktivierung des Hypoxie-induzierbaren Faktors 1α (HIF-1α) (
55
).Die Versteifung proximaler PAs bei PAH-Patienten (
18
,
57
) erhöht den Pulsdruck und die Scherspannung im Lungengefäßsystem. Bedeutsam ist, dass diese Veränderungen der auf die Lungengefäßwand einwirkenden biomechanischen Kräfte entzündungsfördernde Reaktionen in den ECs distaler PAs auslösen (
58
,
59
) und die Aggregation von Immunzellen fördern können (
58
). Dazu gehören die Rekrutierung von Entzündungszellen und die Freisetzung von aus Immunzellen stammenden Zytokinen wie IL-6 (
60
,
61
) und TNF (
62
) sowie bioaktiver Enzyme, einschließlich MMPs (
46
), die wiederum vaskuläre Umbau- und Versteifungsprozesse fördern und so einen fortschreitenden Teufelskreis etablieren können. Ein solches Zusammenspiel zwischen entzündungsbedingten Signalereignissen, die wiederum Wundheilungsprozesse und eine Umgestaltung der extrazellulären Matrix initiieren und letztlich in Gewebefibrose und Narbenbildung gipfeln, ist bei Herz- und systemischen Gefäßerkrankungen gut belegt (
63
–
65
). Bei PH wurde der Ursache-Wirkungs-Zusammenhang zwischen Entzündungssignalen und Gefäßversteifung bislang jedoch weder klinisch noch experimentell untersucht. Daher zielt diese Übersicht darauf ab, bekannte entzündliche Reaktionen bei PH mit Prozessen in Verbindung zu bringen, die mit Gefäßversteifung in Zusammenhang stehen, nämlich mit der Umgestaltung der extrazellulären Matrix und der Gefäßverkalkung, die entweder bei PH oder anderen Gefäßerkrankungen identifiziert wurden, und umgekehrt. Vorgeschlagene Zusammenhänge und relevante Literatur sind in
Tabelle 1
zusammengefasst und werden im Folgenden ausführlich erörtert. Wir möchten damit die potenzielle Relevanz einer pathophysiologischen Achse zwischen Entzündung und PA-Versteifung hervorheben und mechanistische Studien anregen, um diese konzeptionelle Lücke in unserem derzeitigen Verständnis von PH zu schließen. TBELLE 1
Abbildung 1 Vorgeschlagene Rolle der Entzündung bei der PA-Versteifung; Die Entwicklung von PH ist mit einer Entzündungsreaktion im Lungengefäßsystem verbunden, die durch Infiltration von Immunzellen und die Sekretion von Immunfaktoren gekennzeichnet ist. Eine ECM-Versteifung, insbesondere eine proximale großflächige Lungengefäßsklerose, tritt in den frühen Stadien von PH auf und hat einen prognostischen Wert für den Patientenerfolg und die spätere Verkalkung und wird durch Entzündungen verursacht. Eine anhaltende Angiosklerose fördert wiederum eine Entzündungsreaktion, die die Verkalkung und Verdickung der Lungengefäße verschlimmert. PH, pulmonale Hypertonie; ECM, extrazelluläre Matrix; PA, Lungenarterie. Gleichzeitig hat sich im letzten Jahrzehnt die PA-Versteifung als frühes Kennzeichen, Pathomechanismus und Prädiktor von Morbidität und Mortalität bei PH herausgestellt (
18
–
20
). Die Gefäßversteifung, definiert als erhöhter Widerstand der Arterienwand gegen Verformung während des Bluteinflusses, ist eine Folge pathologischer Gefäßumgestaltung, die sowohl in großen proximalen Arterien als auch in kleinen distalen Arterien und Arteriolen auftreten kann. Die mechanischen Folgen dieser Strukturveränderungen sind eine verringerte Compliance in proximalen PAs und ein erhöhter Widerstand gegen den Blutfluss (pulmonaler Gefäßwiderstand, PVR) in distalen PAs (
21
). Die PA-Compliance (PAC) ist wichtig, um den pulsierenden Blutfluss, der über den Windkessel-Effekt in die großen Conduit-Arterien gelangt, in eine nahezu laminare Strömung auf Höhe des distalen pulmonalen Gefäßbaums umzuwandeln. So reduziert PAC die Nachlast des rechten Ventrikels (RV) und hält die Lungendurchblutung über den Herzzyklus hinweg nahezu konstante aufrecht. In Übereinstimmung mit dem Einfluss von PAC auf die RV-Funktion hat die invasive oder nicht-invasive Bewertung von PAC (oder Kapazität) ergeben, dass die PA-Versteifung bei PAH-Patienten ein sensitiver Prädiktor für pathologische RV-Umgestaltung und Mortalität ist (
21
-
24
). Des Weiteren wurde vorgeschlagen, dass eine Versteifung proximaler PAs durch eine Erhöhung der Pulswellengeschwindigkeit und der vom Blut ausgeübten Scherspannung Verletzungen und Umgestaltungen in distalen Gefäßen fördert und so die Pathologie der PH in einer positiven Feedforward-Schleife vorantreibt (
25
). Eine derartige Interdependenz zwischen proximalen und distalen PA-Regionen würde voraussagen, dass pathologische Umgestaltungen in großen und kleinen Gefäßen parallel erfolgen sollten. Tatsächlich wurde in einer Arbeit von Stuart R. Reuben erstmals eine hyperbolische Beziehung zwischen PAC und PVR festgestellt (
26
). Das Produkt aus PAC x PVR ergibt die Resistance-Compliance-Zeit (RC-Zeit), die bei PH-Patienten der WHO-Klasse I (PAH), III (PH aufgrund einer chronischen Lungenerkrankung), IV (chronisch thromboembolische PH) oder V (PH mit unklaren multifaktoriellen Mechanismen) als nahezu konstant und unabhängig von der medikamentösen Therapie angesehen wird (
27
). Interessanterweise ist jedoch bei Patienten mit PH der WHO-Klasse II (PH aufgrund einer Linksherzerkrankung) die RC-Zeit reduziert, d. h. für jeden gegebenen PVR ist die entsprechende PAC niedriger als bei PH-Patienten mit anderen Ursachen. Bemerkenswerterweise ist diese Reduzierung der RC-Zeit auch mit einer Erhöhung der RV-Nachlast verbunden (
27
). Dieser interessante Befund könnte auf andere Pathomechanismen und/oder eine stärkere Versteifung der proximalen PAs bei PH-Patienten mit zugrunde liegender Linksherzerkrankung im Vergleich zu anderen Formen der PH hinweisen; diese Annahme muss jedoch noch streng getestet und mechanistisch untersucht werden.Umgekehrt kann eine mechanische Kommunikation zwischen proximalen PAs und dem distalen Lungengefäßsystem auch die Wiederherstellung der pulmonalvaskulären Homöostase fördern. Hinweise auf einen solchen umgekehrten Umbauprozess stammen aus einigen klinischen Studien bei Patienten mit angeborenen Herzfehlern und PH aufgrund von intrakardialen Links-Rechts-Shunts, die eine Überperfusion der Lunge verursachen. Bei diesen Patienten konnte eine chirurgische Bandierung des PA – durchgeführt mit der Absicht, den proximalen PA vor übermäßigem Druck und Fluss zu schützen – die PH erfolgreich verbessern und in einigen Fällen den vaskulären Umbau in den distalen Arterien umkehren (
28
,
29
).Eine wachsende Zahl von Studien über Techniken zur Abschätzung der Steifigkeit proximaler PAs in vivo zeigt, dass die Abschätzung der PA-Steifigkeit als Prognoseinstrument bei PH vielversprechend ist. Am häufigsten wird die PA-Steifigkeit durch Berechnung der pulmonalarteriellen Kapazität als Verhältnis des Schlagvolumens zum pulmonalen Pulsdruck abgeschätzt, die entweder durch Herzkatheterisierung oder nicht-invasiv durch Echokardiographie ermittelt wird (
20
,
30
–
37
), oder durch Berechnung eines Steifigkeitsindex als Änderung des PA-Drucks (wiederum ermittelt durch Rechtsherzkatheterisierung) geteilt durch die entsprechende Änderung des PA-Durchmessers (bestimmt durch Echtzeit-Bildgebungsverfahren wie kardiale Magnetresonanztomographie) (
18
,
38
).Die Arterienversteifung bei Herz-Kreislauf-Erkrankungen wird hauptsächlich auf eine Umgestaltung der extrazellulären Matrix (ECM) und/oder Verkalkung in der Arterienwand zurückgeführt (
39
–
42
) (
Abbildung 1
). PAH ist insbesondere durch eine Umgestaltung der ECM und eine Verdickung aller drei Schichten der PA-Wand gekennzeichnet (
43
), was letzten Endes die arterielle Compliance reduziert. Die PAs von PAH-Patienten weisen eine erhöhte Ablagerung von interstitiellem Kollagen auf, darunter Kollagen I, Kollagen XIV und Basalmembran-spezifische Kollagene, insbesondere Kollagen IV (
43
–
45
). Darüber hinaus wurde bei PAH-Patienten eine erhöhte Expression anderer ECM-Proteine wie Elastin und Fibronektin oder des matrizellulären ECM-Proteins Tenascin-C durch dedifferenzierte Adventitiafibroblasten beobachtet (
46
). Eine erhöhte Produktion und Ablagerung von ECM-Bestandteilen in PAs wird als adaptive Reaktion auf die erhöhte Verdauung von ECM der Media- und Basalmembran (BM) durch Matrix-Metalloproteinasen (MMPs) angesehen, die bei Patienten mit PAH (
47
) und IPAH (
45
) erhöht sind. Die erhöhte Expression von Kollagen durch Endothelzellen (ECs), glatte Muskelzellen (SMCs) und Adventitiafibroblasten ist mit einer erhöhten Kollagenvernetzung durch Lysyloxidasen (LOXs) verbunden (
48
). Außerdem induzieren proteolytische Enzyme auch den Abbau elastischer Fasern, deren Wiederaufbau trotz erhöhter Elastin-Genexpression aufgrund der komplexen 3D-Mehrkomponentenstruktur dieser Fasern schwierig ist (
49
–
53
). Die PA-Versteifung ergibt sich also als fortschreitendes Ungleichgewicht von Kollagen- gegenüber Elastinfaserkomponenten in der PA-Wand.Eine Gefäßversteifung wird auch auf Gefäßverkalkung (
40
) zurückgeführt, eine pathologische Ablagerung von festen Mineralien in der Intima oder Media der Arterienwände (
54
) (
Abbildung 1
). Wichtig ist, dass die Verkalkung von Lungengefäßen mit einer Transdifferenzierung von SMCs in osteogene Linien in Zusammenhang gebracht wurde, die durch die Aktivität des pro-osteogenen Transkriptionsfaktors RUNX2 (Runt-related transcription factor 2) (
55
) verursacht wird. Eine erhöhte nukleäre Expression von RUNX2 in PA-SMCs aktiviert nicht nur die Expression von verkalkungsbezogenen Biomineralisierungsgenen (
56
), sondern fördert auch die Zellproliferation und Apoptoseresistenz durch Aktivierung des Hypoxie-induzierbaren Faktors 1α (HIF-1α) (
55
).Die Versteifung proximaler PAs bei PAH-Patienten (
18
,
57
) erhöht den Pulsdruck und die Scherspannung im Lungengefäßsystem. Bedeutsam ist, dass diese Veränderungen der auf die Lungengefäßwand einwirkenden biomechanischen Kräfte entzündungsfördernde Reaktionen in den ECs distaler PAs auslösen (
58
,
59
) und die Aggregation von Immunzellen fördern können (
58
). Dazu gehören die Rekrutierung von Entzündungszellen und die Freisetzung von aus Immunzellen stammenden Zytokinen wie IL-6 (
60
,
61
) und TNF (
62
) sowie bioaktiver Enzyme, einschließlich MMPs (
46
), die wiederum vaskuläre Umbau- und Versteifungsprozesse fördern und so einen fortschreitenden Teufelskreis etablieren können. Ein solches Zusammenspiel zwischen entzündungsbedingten Signalereignissen, die wiederum Wundheilungsprozesse und eine Umgestaltung der extrazellulären Matrix initiieren und letztlich in Gewebefibrose und Narbenbildung gipfeln, ist bei Herz- und systemischen Gefäßerkrankungen gut belegt (
63
–
65
). Bei PH wurde der Ursache-Wirkungs-Zusammenhang zwischen Entzündungssignalen und Gefäßversteifung bislang jedoch weder klinisch noch experimentell untersucht. Daher zielt diese Übersicht darauf ab, bekannte entzündliche Reaktionen bei PH mit Prozessen in Verbindung zu bringen, die mit Gefäßversteifung in Zusammenhang stehen, nämlich mit der Umgestaltung der extrazellulären Matrix und der Gefäßverkalkung, die entweder bei PH oder anderen Gefäßerkrankungen identifiziert wurden, und umgekehrt. Vorgeschlagene Zusammenhänge und relevante Literatur sind in
Tabelle 1
zusammengefasst und werden im Folgenden ausführlich erörtert. Wir möchten damit die potenzielle Relevanz einer pathophysiologischen Achse zwischen Entzündung und PA-Versteifung hervorheben und mechanistische Studien anregen, um diese konzeptionelle Lücke in unserem derzeitigen Verständnis von PH zu schließen. TBELLE 1
 Tabelle 1 Entzündungsmediatoren im Zusammenhang mit Gefäßversteifung. Entzündungsbedingte Verdickung der Arterienwände und Umbau der extrazellulären MatrixSowohl die PA-Versteifung als auch entzündliche Reaktionen sind wichtige Merkmale von PH. Während Entzündungen sowohl in Tiermodellen als auch in klinischen Szenarien häufig mit PH in Verbindung gebracht werden, ist über die Rolle von Entzündungen bei der Auslösung der Gefäßumgestaltung bei PH wenig bekannt. Nur wenige Studien haben sich bisher mit der Rolle von Entzündungen bei der Förderung der Produktion von ECM-Komponenten (
154
) befasst, nämlich von Kollagenen (
155
), Fibronektin (
156
) und Tenascin-C (
156
) bei PH. Bei anderen Herz-Kreislauf-Erkrankungen ist der Zusammenhang zwischen Entzündungen und erhöhter Gefäßsteifigkeit jedoch besser charakterisiert: Hier wurde gezeigt, dass entzündliche Prozesse die Arterienversteifung durch eine Vielzahl von Mechanismen fördern, einschließlich der Induktion von endothelialer Dysfunktion und BM-Versteifung, einer erhöhten Proliferation von SMCs (
49
) – was zu einer Verdickung der Arterienwände und verringerter Compliance führt – und einer Umgestaltung und Versteifung der ECM in verschiedenen Segmenten der Arterienwand.Bei PH können erhöhter Druck und hohe pulsierende Strömung als Folge einer verringerten Gefäßcompliance von ECs des pulmonalen Gefäßbetts wahrgenommen werden. Speziell bei Hypoxie-induzierter PH produzieren ECs erhöhte Konzentrationen der inflammatorischen Zytokine IL-1β (
9
) und IL-6 (
9
,
60
) und exprimieren erhöhte Konzentrationen von Immunzell-Adhäsionsmolekülen einschließlich VCAM-1 (Vascular Cell Adhesion Molecule-1), ICAM-1 (Intercellular Adhesion Molecule-1) und P-Selectin (
9
). Gleichzeitig reagieren andere vaskuläre Zellen wie SMCs und Fibroblasten auf biomechanische Signale mit einer veränderten Sekretion von Immunfaktoren, darunter inflammatorische Zytokine wie MCP-1 (Monocyte Chemoattractant Protein-1), Stroma Cell-Derived Factor 1 und CCR5 (
71
) (
Tabelle 1
). Diese Entzündungsmediatoren können wiederum eine Umgestaltung und Versteifung der PA bewirken (
9
,
71
,
157
). Während die Zusammenhänge zwischen erhöhter Entzündung und PA-Umgestaltung bei PH bislang noch wenig verstanden sind, werden wir im Folgenden bestehende Zusammenhänge zwischen wichtigen Entzündungssignalen und Gefäßversteifung bei systemischen Herz-Kreislauf-Erkrankungen darstellen, mit dem Ziel, dieses Wissen in ein erweitertes Verständnis der möglichen Rolle von Entzündungen bei der PA-Versteifung bei PH umzusetzen.Mehrere wichtige Entzündungssignale induzieren eine PA-Umgestaltung, indem sie das Verhalten und die Funktion von ECs und SMCs bei PH dysregulieren, was letztendlich zu einer Verdickung und Versteifung der Arterienwände führt. Unter diesen wurden erhöhte IL-6- und TNF-Werte im Plasma, in der Lunge, in den Lungenarterien und -venen sowie in PA-ECs sowohl bei Patienten als auch bei Tiermodellen verschiedener PH-Gruppen festgestellt (
Tabelle 1
). Bei PAH-Patienten (
60
) und in PH-LHD-Rattenmodellen (
61
,
81
) trägt IL-6 zur PA-Umgestaltung bei, indem es eine Verdickung der Mediawand über SMC-Proliferation und Muskularisierung des distalen Pulmonalarterienbaums aufgrund der Migration von SMCs in präkapilläre Arteriolen (
60
,
61
,
81
) induziert (
Tabelle 2
;
Abbildung 2
), was möglicherweise die arterielle Compliance durch erhöhte Wandverdickung beeinträchtigt. In der pulmonalen Adventitia aktivieren Fibroblasten rekrutierte Makrophagen durch parakrine IL-6-Signalgebung und initiieren damit einen proinflammatorischen und profibrotischen Phänotyp, der mit einer erhöhten Entzündungsreaktion und Gefäßumgestaltung bei PH verbunden ist (
7
). Insbesondere ist IL-6 ein sensitiver Marker für systemische Entzündungen bei Herz-Kreislauf-Erkrankungen (
60
,
88
). Bei rheumatoider Arthritis (
164
) und akutem ischämischem Schlaganfall (
165
) waren erhöhte IL-6-Konzentrationen im Patientenserum mit einer anhand der Pulswellengeschwindigkeit abgeschätzten Aortenversteifung verbunden, die durch therapeutische Infusionen des Anti-IL-6-Rezeptor-Antikörpers Tocilizumab signifikant reduziert werden konnte (
164
). TBELLE 2
Tabelle 1 Entzündungsmediatoren im Zusammenhang mit Gefäßversteifung. Entzündungsbedingte Verdickung der Arterienwände und Umbau der extrazellulären MatrixSowohl die PA-Versteifung als auch entzündliche Reaktionen sind wichtige Merkmale von PH. Während Entzündungen sowohl in Tiermodellen als auch in klinischen Szenarien häufig mit PH in Verbindung gebracht werden, ist über die Rolle von Entzündungen bei der Auslösung der Gefäßumgestaltung bei PH wenig bekannt. Nur wenige Studien haben sich bisher mit der Rolle von Entzündungen bei der Förderung der Produktion von ECM-Komponenten (
154
) befasst, nämlich von Kollagenen (
155
), Fibronektin (
156
) und Tenascin-C (
156
) bei PH. Bei anderen Herz-Kreislauf-Erkrankungen ist der Zusammenhang zwischen Entzündungen und erhöhter Gefäßsteifigkeit jedoch besser charakterisiert: Hier wurde gezeigt, dass entzündliche Prozesse die Arterienversteifung durch eine Vielzahl von Mechanismen fördern, einschließlich der Induktion von endothelialer Dysfunktion und BM-Versteifung, einer erhöhten Proliferation von SMCs (
49
) – was zu einer Verdickung der Arterienwände und verringerter Compliance führt – und einer Umgestaltung und Versteifung der ECM in verschiedenen Segmenten der Arterienwand.Bei PH können erhöhter Druck und hohe pulsierende Strömung als Folge einer verringerten Gefäßcompliance von ECs des pulmonalen Gefäßbetts wahrgenommen werden. Speziell bei Hypoxie-induzierter PH produzieren ECs erhöhte Konzentrationen der inflammatorischen Zytokine IL-1β (
9
) und IL-6 (
9
,
60
) und exprimieren erhöhte Konzentrationen von Immunzell-Adhäsionsmolekülen einschließlich VCAM-1 (Vascular Cell Adhesion Molecule-1), ICAM-1 (Intercellular Adhesion Molecule-1) und P-Selectin (
9
). Gleichzeitig reagieren andere vaskuläre Zellen wie SMCs und Fibroblasten auf biomechanische Signale mit einer veränderten Sekretion von Immunfaktoren, darunter inflammatorische Zytokine wie MCP-1 (Monocyte Chemoattractant Protein-1), Stroma Cell-Derived Factor 1 und CCR5 (
71
) (
Tabelle 1
). Diese Entzündungsmediatoren können wiederum eine Umgestaltung und Versteifung der PA bewirken (
9
,
71
,
157
). Während die Zusammenhänge zwischen erhöhter Entzündung und PA-Umgestaltung bei PH bislang noch wenig verstanden sind, werden wir im Folgenden bestehende Zusammenhänge zwischen wichtigen Entzündungssignalen und Gefäßversteifung bei systemischen Herz-Kreislauf-Erkrankungen darstellen, mit dem Ziel, dieses Wissen in ein erweitertes Verständnis der möglichen Rolle von Entzündungen bei der PA-Versteifung bei PH umzusetzen.Mehrere wichtige Entzündungssignale induzieren eine PA-Umgestaltung, indem sie das Verhalten und die Funktion von ECs und SMCs bei PH dysregulieren, was letztendlich zu einer Verdickung und Versteifung der Arterienwände führt. Unter diesen wurden erhöhte IL-6- und TNF-Werte im Plasma, in der Lunge, in den Lungenarterien und -venen sowie in PA-ECs sowohl bei Patienten als auch bei Tiermodellen verschiedener PH-Gruppen festgestellt (
Tabelle 1
). Bei PAH-Patienten (
60
) und in PH-LHD-Rattenmodellen (
61
,
81
) trägt IL-6 zur PA-Umgestaltung bei, indem es eine Verdickung der Mediawand über SMC-Proliferation und Muskularisierung des distalen Pulmonalarterienbaums aufgrund der Migration von SMCs in präkapilläre Arteriolen (
60
,
61
,
81
) induziert (
Tabelle 2
;
Abbildung 2
), was möglicherweise die arterielle Compliance durch erhöhte Wandverdickung beeinträchtigt. In der pulmonalen Adventitia aktivieren Fibroblasten rekrutierte Makrophagen durch parakrine IL-6-Signalgebung und initiieren damit einen proinflammatorischen und profibrotischen Phänotyp, der mit einer erhöhten Entzündungsreaktion und Gefäßumgestaltung bei PH verbunden ist (
7
). Insbesondere ist IL-6 ein sensitiver Marker für systemische Entzündungen bei Herz-Kreislauf-Erkrankungen (
60
,
88
). Bei rheumatoider Arthritis (
164
) und akutem ischämischem Schlaganfall (
165
) waren erhöhte IL-6-Konzentrationen im Patientenserum mit einer anhand der Pulswellengeschwindigkeit abgeschätzten Aortenversteifung verbunden, die durch therapeutische Infusionen des Anti-IL-6-Rezeptor-Antikörpers Tocilizumab signifikant reduziert werden konnte (
164
). TBELLE 2
 Tabelle 2 Mögliche Zusammenhänge zwischen Faktoren, die mit der PA-Versteifung in Zusammenhang stehen, und Immunreaktionen bei PH. ABILDUNG 2
Tabelle 2 Mögliche Zusammenhänge zwischen Faktoren, die mit der PA-Versteifung in Zusammenhang stehen, und Immunreaktionen bei PH. ABILDUNG 2
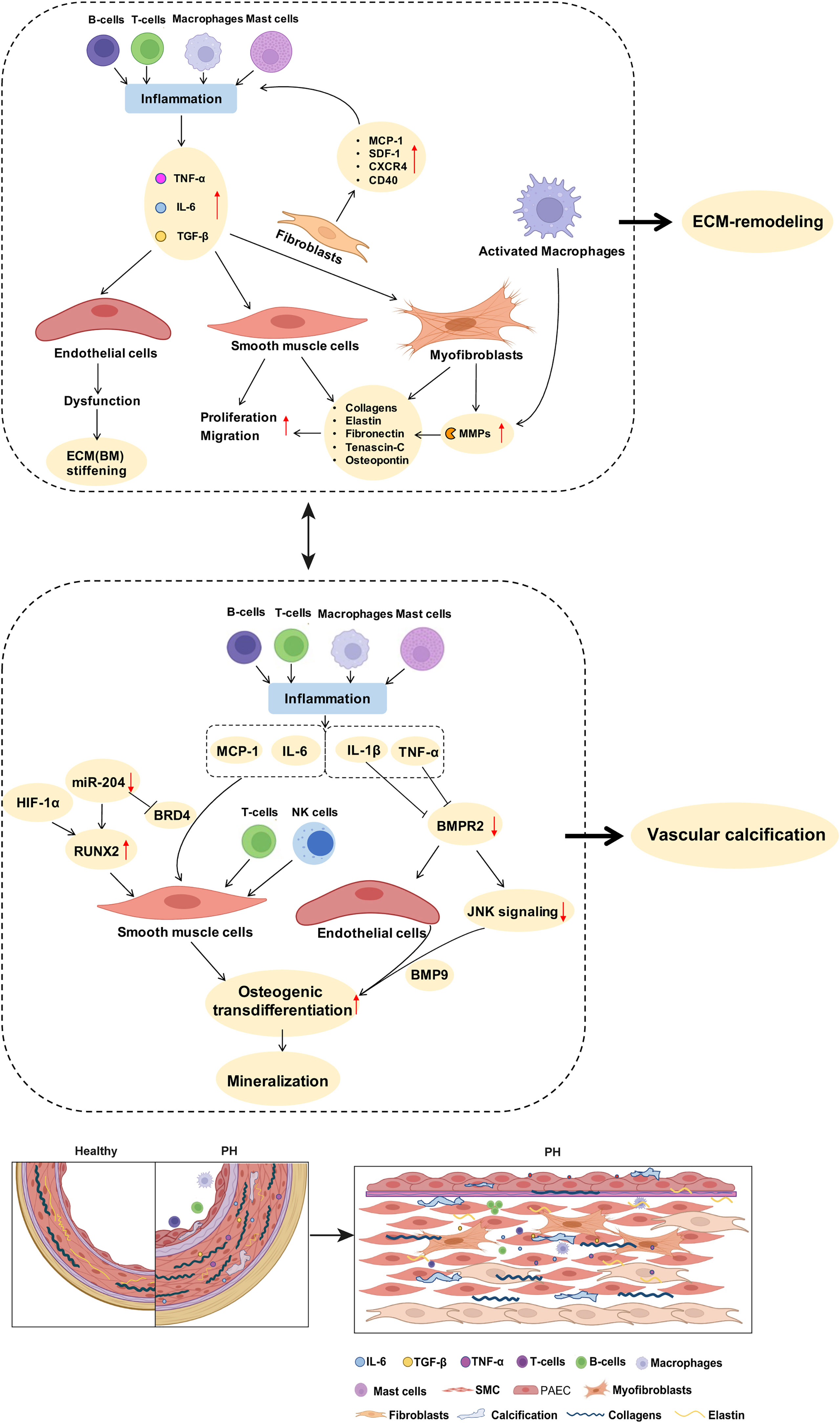 Abbildung 2 Mögliche Verbindungen zwischen Entzündungsmediatoren und Mechanismen der Umgestaltung der pulmonalarteriellen ECM und der Gefäßverkalkung bei PH. Wie im Manuskripttext ausführlich beschrieben, ist die perivaskuläre Ansammlung von Immunzellen ein charakteristisches Merkmal von PH. Entzündungszellen wie Makrophagen produzieren MMPs, die den Abbau und die Umgestaltung der ECM fördern. Entzündungszytokine wie IL-6 und TGF-β treiben die Proliferation von PA-SMCs voran. Die Stimulierung von Fibroblasten durch Entzündungsmediatoren erhöht die Expression von Kollagenen, Elastin und Fibronektin, was die PA-Steifheit weiter fördert. Aktivierte Immunzellen und Entzündungsmediatoren fördern die SMC-Transdifferenzierung und erhöhen die Expression von Biomineralisierungsgenen, wodurch die Gefäßverkalkung gefördert wird. Die Herunterregulierung von BMPR2, insbesondere als Reaktion auf den Entzündungsfaktor TNF, fördert die Mesenchymisierung von Endothelzellen und kann als solche zur Entwicklung einer pulmonalarteriellen Gefäßverkalkung beitragen. Eine detaillierte Erörterung der vorgeschlagenen Signalwege findet sich im Manuskripttext. ECM, extrazelluläre Matrix; MMPs, Matrix-Metalloproteinasen; SMC, glatte Muskelzellen; PH, pulmonale Hypertonie; PA, Pulmonalarterie; BM, Basalmembran. In ähnlicher Weise wurde gezeigt, dass erhöhte TNF-Werte in Nagetiermodellen von PAH und PH-LHD zu einer erhöhten PA EC- und SMC-Proliferation und einer Verdickung der medialen Wand führen (
61
,
166
), was auf eine unterdrückte BMPR-II-Signalgebung bei PAH zurückgeführt wurde (
166
). Aufgrund seiner Wirkung auf die SMC-Hyperplasie kann TNF auch die PA-Versteifung bei PH fördern; direkte Zusammenhänge zwischen TNF-Werten und PA-Versteifung bei PH müssen jedoch noch nachgewiesen werden. Bei anderen kardiovaskulären und entzündlichen Erkrankungen, z. B. Arteriosklerose, ist TNF ein etablierter Schlüsselmediator der Gefäßumgestaltung (
61
,
62
). Bei Patienten mit entzündlichen Arterienerkrankungen, nämlich rheumatoider Arthritis, ankylosierender Spondylitis und Psoriasis-Arthritis, die eine Anti-TNF-Behandlung mit Adalimumab, Etanercept oder Infliximab erhielten, wurde im Vergleich zu unbehandelten Kontrollpersonen eine Reduktion der Aortensteifigkeit gemessen anhand der Pulswellengeschwindigkeit und des Augmentationsindex festgestellt (
107
,
167
). Daher könnte die pharmakologische Hemmung von Entzündungsmediatoren wie IL-6 und TNF bei PH möglicherweise die Proliferation pulmonaler Gefäßzellen und die PA-Verdickung reduzieren und könnte daher eine gezielte Therapie für die PA-Versteifung darstellen.Darüber hinaus können proinflammatorische Mediatoren bei Herz-Kreislauf-Erkrankungen eine Gefäßversteifung durch eine erhöhte Produktion von ECM-Komponenten, nämlich fibrillären und nichtfibrillären Kollagenen und Fibronektin durch residente Gefäßzellen induzieren (
168
). Nach einem Herzinfarkt sowie bei ischämischer und nicht-ischämischer Herzinsuffizienz induzieren proinflammatorische Mediatoren wie TGF-β (
74
,
169
) und IL-1β (
170
) die Umwandlung von Fibroblasten in Myofibroblasten, die reichlich ECM-Proteine produzieren können (
168
) (
Tabellen 1
,
2
;
Abbildung 2
). Bei PH tragen adventitiale Myofibroblasten durch die Produktion struktureller ECM-Komponenten wie Kollagene, Elastin, Fibronektin und dynamischer ECM-Bestandteile wie Tenascin-C und Osteopontin (
43
,
46
,
74
) (
Tabelle 2
;
Abbildung 2
)
zur Umgestaltung und Versteifung der PA bei (46). Tenascin-C und Osteopontin wiederum erhöhen die Proliferation von Fibroblasten und SMC, was zur Umwandlung von Myofibroblasten und Mediaverdickung und damit zur Gefäßversteifung beiträgt (
43
,
46
,
171
) (
Abbildung 2
). In die pulmonale Adventitia rekrutierte aktivierte Makrophagen können ECM-Proteine wie Kollagen Typ I exprimieren und so zur ECM-Versteifung bei PH beitragen (
172
). In Tiermodellen mit MCT-induzierter PH wurde auch eine Hochregulierung der NADPH-Oxidase 4 (Nox4) in der pulmonalen Adventitia festgestellt, wo sie die TGF-β-vermittelte Expression von Matrixkollagenen durch Adventitiafibroblasten und damit die ECM-Versteifung fördert (
172
). In ähnlicher Weise wurde in einem Tiermodell mit chronischer hypoxischer PH eine erhöhte Kollagenablagerung durch residente Fibroblasten in der Adventitia festgestellt, was zu einer dickeren und steiferen Arterienwand führte (
172
–
174
). Um unlösliche, starre Fasern zu bilden, werden überschüssige fibrilläre Kollagene dann durch Vernetzungsenzyme weiter vernetzt (
43
,
175
). Insbesondere eine erhöhte Expression von LOX in SMCs und eine Expression des Lysyloxidase-ähnlichen Enzyms (LOXL) in Adventitiafibroblasten führt zu einer erhöhten Kollagenvernetzung und PA-Versteifung bei PAH (
176
). Darüber hinaus weisen Adventitiafibroblasten per se einen proinflammatorischen Phänotyp bei PH auf, einschließlich der Rekrutierung und Aktivierung von Adventitiamakrophagen (
7
) und zur Produktion proinflammatorischer Marker wie der Chemokine MCP-1, SDF-1, RANTES/CCR5, CCR7, CXCR4 und der Kostimulationsmoleküle CD40 und CD40L (
7
,
71
). Diese sekretorische Aktivität kann wiederum eine Rückkopplungsschleife erzeugen, die weitere Entzündungen und damit eine Umgestaltung der ECM auslöst.Neben erhöhten Konzentrationen zirkulierender Entzündungsmediatoren induziert erhöhter mPAP-Spiegel bei PH auch die Aktivierung des proinflammatorischen NF-κB-Signalwegs in PA ECs und SMCs (
58
,
133
,
157
) (
Abbildung 2
). Basierend auf Studien zu systemischen Herz-Kreislauf-Erkrankungen erweist sich eine solche Aktivierung von NF-κB als potenziell wichtiger Schritt bei der PA-Versteifung. So wurde gezeigt, dass nukleäres NF-κB die Expression von Aortenkollagen Typ I in einem Mausmodell für Typ-2-Diabetes erhöht, was zu einer Aortenversteifung führt, die ex vivo durch Druckmyographie gemessen wurde (
177
). Interessanterweise wurden diese Effekte durch eine NF-κB-abhängige Überexpression von RUNX2 vermittelt, einem wichtigen Transkriptionsfaktor, der nicht nur für die ECM-Umgestaltung [durch erhöhte Expression von ECM-Kollagenen durch SMCs (
177
)] relevant ist, sondern auch im Zusammenhang mit der Gefäßverkalkung (
55
,
177
) (wie unten erläutert) (
Abbildung 2
). Es lässt sich spekulieren, dass die Aktivierung von NF-κB ähnliche Effekte bei PH haben und so zur PA-Versteifung durch ECM-Umgestaltung und Gefäßverkalkung beitragen könnte.Der entzündungsbedingten Überproduktion von ECM-Komponenten bei Herz-Kreislauf-Erkrankungen steht ein erhöhter proteolytischer Abbau der ECM infolge eines parallelen Anstiegs von MMPs gegenüber (
46
). Bei PH sezernieren aktivierte Makrophagen und Myofibroblasten in der Adventitia MMPs, insbesondere MMP-2 (
154
,
162
), MMP-9 (
6
,
162
), MMP-10 (
47
) und MMP-19 (
6
,
154
), während Gewebeinhibitoren von Metalloproteinasen (TIMPs) herunterreguliert zu sein scheinen (
46
,
162
) (
Tabelle 2
;
Abbildung 2
). MMP-2 (
50
) und MMP-9 (
49
) bauen Elastin ab und verringern dadurch die Gefäßcompliance, was zu einer Arterienversteifung führt (
49
–
52
). Darüber hinaus erleichtert der Abbau elastischer Fasern und anderer ECM-Bestandteile wie BM-Kollagen, interstitieller Kollagene, Fibronektin und verschiedener Proteoglykane durch MMPs die Migration von Adventitiafibroblasten und Myofibroblasten in die Media und Intima, was wiederum die PA-Versteifung und Gefäßstenose fördert (
46
,
154
) (
Tabelle 2
). In ähnlicher Weise wird die Neointimalbildung durch erhöhte Proliferation und Migration von SMC aus der Media in die Intimabereiche der Arterienwand durch MMP-regulierten ECM-Abbau erleichtert (
52
,
178
) und fördert Gefäßstenose und -versteifung (
178
). Produkte der ECM-Proteolyse – die Matrikine [kürzlich ausführlich besprochen von Mutgan et al. (
179
)] – können wiederum als entzündungsfördernde Mediatoren dienen, die die Entzündung verstärken und so eine weitere positive Rückkopplungsschleife erzeugen können (
43
). Darüber hinaus ermöglicht der Abbau der ECM, dass zirkulierende Serumfaktoren in das Medium gelangen und die Produktion von Serinelastase durch SMCs stimulieren (
178
). Diese Serinelastasen unterstützen den Abbau von Elastin und die Freisetzung aktivierter Wachstumsfaktoren wie Fibroblastenwachstumsfaktor (FGF) und TGF-β, die wiederum die Produktion von Kollagen, Fibronektin und Tenascin-C durch SMCs und Fibroblasten erhöhen (
43
) – was wiederum die PA-Versteifung fördert (
Tabelle 2
). Bei anderen Herz-Kreislauf-Erkrankungen wie ischämischer Herzinsuffizienz sezernieren Immunzellen wie Makrophagen, Lymphozyten und Mastzellen MMPs, die die vaskuläre und kardiale ECM als Reaktion auf mechanische Belastung umgestalten (
168
). Bei Arteriosklerose waren erhöhte Konzentrationen von MMP-2 und MMP-9 mit einer erhöhten Arteriensteifigkeit und einem erhöhten Risiko für Herz-Kreislauf-Erkrankungen verbunden, was auf ihre Fähigkeit zurückgeführt wurde, die elastischen Schichten in den Arterien abzubauen (
180
,
181
). Dementsprechend reduziert der MMP-2-Knockdown die Arteriensteifigkeit der Halsschlagadern bei Mäusen, indem er den Elastinabbau im Gewebe verringert (
182
).So können sich die Aktivierung von Immunzellen und Entzündungswegen sowie die Verdickung der Arterienwände und die Umgestaltung der extrazellulären Matrix (ECM) gegenseitig stimulieren. Die gezielte Beeinflussung entzündlicher Prozesse bei Herz-Kreislauf-Erkrankungen, z. B. Aortenaneurysmen, hat sich als vorteilhaft für Schlüsselmechanismen der Umgestaltung der extrazellulären Matrix (ECM) erwiesen, wie z. B. Elastinabbau, MMP-Expression und Makrophageninfiltration (
183
). Ein besseres Verständnis der spezifischen Akteure und molekularen Wege, die an dieser gegenseitigen Interaktion beteiligt sind, kann neue und möglicherweise personalisierte Ziele für die zukünftige PH-Therapie aufzeigen.Verkalkung und Entzündung der LungenarterienBiologisch induzierte Mineralisierung ist ein integraler Bestandteil der menschlichen Physiologie und Gewebehomöostase. Dabei werden extra- und intrazelluläre Mechanismen eingesetzt, um die Bildung, das Wachstum und die Lokalisierung der abgelagerten Mineralien zu steuern. Unter Krankheitsbildern können diese Prozesse durch Veränderungen des lokalen oder globalen Kalziummilieus, DNA-Schäden, Stress des endoplasmatischen Retikulums, oxidativen Stress oder Stoffwechselstörungen – also Prozesse, die häufig mit Entzündungsreaktionen einhergehen – aus dem Gleichgewicht geraten und letztendlich zu pathologischer Verkalkung von Gewebe oder Blutgefäßen führen (
184
,
185
). Mechanistisch gesehen führen diese Faktoren zu (oder werden begleitet von) einer phänotypischen Umwandlung verschiedener Zelltypen in Osteoprogenitorzellen durch eine De-novo- bzw. erhöhte Expression des potenten Transkriptionsaktivators RUNX2, der die Expression nachgeschalteter, kalzifizierungsfördernder Proteine wie alkalischer Phosphatase auslöst (
186
–
188
). Im Vergleich zu systemischen Arterien wird die Gefäßverkalkung der PA kaum thematisiert, obwohl sie tatsächlich ein häufiges Merkmal bei Patienten mit schwerer, lang anhaltender PH (
189
), fortgeschrittener PH und PH mit chronischem Nierenversagen (
190
) oder terminaler Niereninsuffizienz (
191
) ist. Tatsächlich sagt der Nachweis einer Verkalkung der peripheren PA mittels Computertomographie (CT) (
192
) den Langzeitverlauf bei PH (
193
) und bei Patienten mit Vorhofseptumdefekt und Eisenmenger-Syndrom (
194
) voraus.Im Zusammenhang mit PAH wurde einer microRNA-204-abhängigen Hochregulierung von RUNX2 eine entscheidende Rolle bei der Regulierung der PA-Kalzifizierung zugeschrieben, die wiederum HIF-1α aktiviert, was zu einer Hyperproliferation von PA-SMCs, einer Resistenz gegen Apoptose und einer anschließenden Transdifferenzierung in osteoblastenähnliche Zellen führt (
55
). Eine zweite Studie berichtete, dass Hypoxie-induzierte zirkuläre RNA CDR1 die osteogene Transdifferenzierung menschlicher PA-SMCs fördert, indem sie microRNA-7-5p aufsaugt und infolgedessen ihre nachgeschalteten Zielmoleküle Calcium/Calmodulin-abhängige Kinase II-delta (CAMK2D) und Calponin 3 (CNN3) hochreguliert (
195
). Drittens steht die PA-Verkalkung in Zusammenhang mit Hypoxie, da Hypoxie die Expression der in den Granula von T-Lymphozyten und natürlichen Killerzellen gespeicherten Serinprotease Granzym B verringert, was die speichergesteuerten Calciumkanäle (SOCCs) als Hauptquelle des Minerals Calcium hemmt, indem es nicht-kanonische Wnt-Signale in SMCs abschwächt und so die Verkalkung der PA verstärkt (
141
). Unabhängig vom zugrundeliegenden Signalweg erhöht die Verkalkung letztlich die Gefäßsteifigkeit und verringert die Compliance der Pulmonalarterienwand, was ein Ausdruck der schlechten Prognose bei PH ist (
21
).Es wurde gezeigt, dass Entzündungssignale – wie sie bei PH auftreten – im systemischen Gefäßsystem vaskuläre Verkalkungsprozesse regulieren. Insbesondere fördert TNF die osteogene Differenzierung und Verkalkung boviner aortaler SMCs, indem es die Expression von Osteoblastenmarkern wie osteoblastenspezifischem Faktor 2 (Osf2), Aktivatorprotein 1 (AP1) und cAMP-responsivem Element-Bindungsprotein (CREB) durch Aktivierung der cAMP-Signalgebung induziert (
104
). Ebenso verursachte die Behandlung aortaler SMCs mit IL-1β oder IL-6 eine dosisabhängige Erhöhung der alkalischen Phosphataseaktivität und eine erhöhte Zellmineralisierung in vitro (
196
). Interessanterweise wird die Expression der inflammatorischen Zytokine IL-6, TNF und MCP-1 in verschiedenen Geweben epigenetisch durch das Bromodomain-Protein 4 (BRD4) reguliert (
86
), das die Chromatinlandschaft moduliert und die Genexpression aktiviert, indem es Transkriptionsfaktoren an Genpromotoren und/oder Superenhancern aufbaut. Insbesondere ist BRD4 in PA-SMCs von PAH-Patienten und in Lungen oder distalen PAs von Ratten-PH-Modellen hochreguliert und wird posttranskriptionell durch microRNA-204 reguliert (
87
), das gleichzeitig an der PA-Verkalkung beteiligt ist (
55
), wodurch eine zusätzliche epigenetische Verbindung zwischen Entzündung und Gefäßverkalkung hergestellt wird. Wichtiger noch: Es wurde gezeigt, dass der RUNX2-Genpromotor sowohl während der Osteoblastendifferenzierung (
197
) als auch bei Krebs (
198
) unter direkter Kontrolle von BRD4 steht, was nahelegt, dass BRD4 als „Hauptregulator“ sowohl der Entzündung als auch der Gefäßverkalkung gleichzeitig dienen kann. In Übereinstimmung mit dieser Annahme schwächte die BRD4-Hemmung die Umgestaltung der Lungen- und Koronararterien bei experimenteller PH ab, und dieser Schutzeffekt war mit verringerten IL-6- und MCP-1-Spiegeln verbunden (
199
,
200
).Obwohl es nur wenige Studien gibt, die Verkalkung und Entzündung bei PH in Zusammenhang bringen, sind Zytokine an der Regulierung der Verkalkung in den extrapulmonalen Gefäßen beteiligt. Wichtig ist, dass die Gefäßverkalkung auch eng mit der Umgestaltung und Versteifung der ECM verknüpft zu sein scheint (
201
), da die Mineralisierung der SMC direkt mit der Produktion von Kollagen I und dem Abbau von Fibronektin und Elastin korreliert, während letzteres Gerüste für die Kalziumeinlagerung bildet (
201
–
203
). Diese Erkenntnisse legen nahe, dass vorgelagerte Entzündungen durch die Umgestaltung der ECM auch die Gefäßverkalkung fördern können.Pulmonalarterieller endothelialer-mesenchymaler Übergang und EntzündungWährend sich unsere Untersuchung von Gefäßverkalkungsprozessen weitgehend auf SMCs konzentriert hat, ist es wichtig zu bedenken, dass auch ECs beteiligt sind. Bei verschiedenen Herz-Kreislauf-Erkrankungen verlieren ECs ihre charakteristische Morphologie und verändern sich hin zu einem mesenchymalen Phänotyp (
204
), ein Prozess, der als endothelial-mesenchymaler Übergang (EndoMT) bezeichnet wird und insbesondere durch Entzündungen moduliert wird. Insbesondere wurde gezeigt, dass entzündliche Zytokine wie IL-1β oder TNF EndoMT in PA ECs induzieren. Im Gegenzug beginnen diese EndoMT-Zellen, entzündliche Zytokine wie IL-4, IL-6, IL-8, IL-13 und TNF in viel höheren Konzentrationen als normale PA ECs abzusondern (
205
), wodurch eine potenziell bösartige Feedforward-Schleife entsteht. Im Einklang mit der Theorie der entzündungsbedingten EndoMT bei PH zeigte sich, dass die Aktivierung des proinflammatorischen NF-κB-Signalwegs in einem Mausmodell der durch Monocrotalin (MCT) induzierten PH zu einer Hochregulierung von miR-130a führte, was zu einem Verlust des Knochenmorphogenetischen Proteinrezeptors Typ 2 (BMPR2), einer erhöhten Expression von HMGA1 (High Mobility Group AT-hook 1) und schließlich zu EndoMT in mikrovaskulären endothelialen Zellen der Lunge führte (
206
). Es ist wichtig hervorzuheben, dass EndoMT zwar ausführlich in pulmonalen und systemischen endokrinen Zellen (ECs), die Entzündungsmediatoren ausgesetzt waren, in vitro dokumentiert wurde , das Ausmaß und die Relevanz von EndoMT in vivo in jüngsten Studien unter Verwendung von Technologien zur Abstammungsverfolgung jedoch weiterhin umstritten sind: Durch Verwendung von doppelt transgenen Mäusen, die in allen ECs stabil grünes fluoreszierendes Protein (GFP) exprimieren, konnten Suzuki und Kollegen GFP in 14,3 ± 1,8 % der mesenchymalen (CD144 - CD45 - CD326 - ) Zellen nachweisen, was auf eine substantielle EndoMT hinweist (
207
). In ähnlicher Weise zeigte die Endothel-Linienverfolgung unter Verwendung transgener vaskulärer Endothel-Cadherin-Cre-Rekombinase oder Tie-2 Cre-Mäuse, die mit mTomato/mGreen Fluoreszenzprotein-Doppelfluoreszenz-Cre-Reportermäusen gekreuzt wurden, reichlich endotheliale Linien-markierte Zellen in der Neointima, wo sie nach Induktion von PH durch Monocrotalin-Pyrrol glatte Muskel-α-Aktin und glatte Muskel-Myosin-Schwerketten exprimierten (
208
). Eine kürzlich durchgeführte Linienverfolgungsstudie in chronischen Hypoxie- und allergeninduzierten Modellen der Lungengefäßumgestaltung zeigte jedoch die Beibehaltung endothelialer Linien-spezifischer Marker-Expressionsprofile ohne Anzeichen einer Zelltypumwandlung (
209
). Insbesondere steht die Erkennung einer eingeschränkten oder partiellen EndoMT nicht unbedingt im Widerspruch zu ihrer potenziellen funktionellen Relevanz bei der PA-Versteifung, sondern legt lediglich nahe, dass diese Relevanz möglicherweise eher mit der Freisetzung proliferativer, hypertropher und profibrotischer Signale – also Mediatoren von Prozessen, die letztlich die PA-Versteifung fördern – durch partielle EndoMT-Zellen zusammenhängt als mit der tatsächlichen Bildung einer signifikanten mesenchymalen Zellmasse über diesen Mechanismus. Tatsächlich wird eine ähnliche Rolle zunehmend für den epithelial-mesenchymalen Übergang bei Gewebefibrose erkannt (
210
).Darüber hinaus kann EndoMT Entzündungen mit Gefäßverkalkung und damit mit PA-Versteifung bei PH in Verbindung bringen. Insbesondere Studien an aortischen endothelialen Zellen (ECs) zeigen, dass inflammatorische Zytokine wie TNF und IL-1β EndoMT modulieren und die Expression der BMPR2- und JNK-Signalgebung herunterregulieren, wodurch ECs für BMP9-induzierte osteogene Differenzierung sensibilisiert werden, die in Mineralisierung gipfelt (
141
). Ähnliche Regulationsmechanismen können die Verkalkung von PA-ECs bei verschiedenen Arten von PH vorantreiben, und PAH-Patienten mit BMPR2-Mutationen oder Beeinträchtigungen des BMP-Signalwegs (
104
) wären in diesem Szenario voraussichtlich besonders anfällig, da eine Beeinträchtigung der BMPR2-Signalgebung mit EndoMT in Zusammenhang steht (
211
). Abstammungsverfolgungsstudien im systemischen Kreislauf unterstützen eine Rolle von EndoMT bei der Gefäßverkalkung. Sie zeigen beispielsweise, dass eine Untergruppe von Endokardzellen eine endokardial-mesenchymale Transition durchlaufen kann, die zur Verkalkung der Herzklappen von Mäusen und Menschen führt (
212
), oder dass vaskuläre endokardiale Zellen in osteogene Zellen übergehen können (
213
), was durch Hemmung der Glykogensynthase-Kinase 3 (GSK3) verhindert werden kann (
214
). Die Rolle von EndoMT (oder partiellem EndoMT) bei der Gefäßverkalkung im Lungenkreislauf und im Zusammenhang mit PH wurde bisher jedoch nicht untersucht.Mögliche klinische RelevanzWährend sich aktuelle PH-Therapien (z. B. Prostazykline, Phosphodiesterasehemmer, Kalziumkanalblocker, Endothelin-Rezeptorantagonisten oder Stimulatoren der löslichen Guanylatcyclase) in erster Linie auf die Linderung der Vasokonstriktion als symptomatischen Ansatz konzentrieren (
215
), besteht das langfristige therapeutische Ziel darin, sich auf die Mechanismen zu konzentrieren, die für den Ausbruch und das Fortschreiten der Krankheit verantwortlich sind, darunter Gefäßumbau und Entzündung (
215
). In dieser Hinsicht könnte das gezielte Angreifen der Immun-PA-Versteifungsachse eine besonders vielversprechende Strategie sein, angesichts der prädiktiven und pathomechanistischen Rolle der PA-Versteifung bei PH und des Arsenals immunmodulatorischer Therapien, die sich bereits im klinischen Einsatz oder in der Entwicklung befinden. Im systemischen Kreislauf haben sich entzündungshemmende Therapien als vielversprechend bei der Verringerung der Arterienversteifung bei entzündlichen Arterienerkrankungen wie rheumatoider Arthritis oder Psoriasis-Arthritis erwiesen (
167
). Insbesondere TNF-Antagonisten wie Adalimumab, Etanercept oder Infliximab sind etablierte entzündungshemmende Therapien bei (Auto-)Immunerkrankungen (
216
), die die Aortensteifigkeit
Abbildung 2 Mögliche Verbindungen zwischen Entzündungsmediatoren und Mechanismen der Umgestaltung der pulmonalarteriellen ECM und der Gefäßverkalkung bei PH. Wie im Manuskripttext ausführlich beschrieben, ist die perivaskuläre Ansammlung von Immunzellen ein charakteristisches Merkmal von PH. Entzündungszellen wie Makrophagen produzieren MMPs, die den Abbau und die Umgestaltung der ECM fördern. Entzündungszytokine wie IL-6 und TGF-β treiben die Proliferation von PA-SMCs voran. Die Stimulierung von Fibroblasten durch Entzündungsmediatoren erhöht die Expression von Kollagenen, Elastin und Fibronektin, was die PA-Steifheit weiter fördert. Aktivierte Immunzellen und Entzündungsmediatoren fördern die SMC-Transdifferenzierung und erhöhen die Expression von Biomineralisierungsgenen, wodurch die Gefäßverkalkung gefördert wird. Die Herunterregulierung von BMPR2, insbesondere als Reaktion auf den Entzündungsfaktor TNF, fördert die Mesenchymisierung von Endothelzellen und kann als solche zur Entwicklung einer pulmonalarteriellen Gefäßverkalkung beitragen. Eine detaillierte Erörterung der vorgeschlagenen Signalwege findet sich im Manuskripttext. ECM, extrazelluläre Matrix; MMPs, Matrix-Metalloproteinasen; SMC, glatte Muskelzellen; PH, pulmonale Hypertonie; PA, Pulmonalarterie; BM, Basalmembran. In ähnlicher Weise wurde gezeigt, dass erhöhte TNF-Werte in Nagetiermodellen von PAH und PH-LHD zu einer erhöhten PA EC- und SMC-Proliferation und einer Verdickung der medialen Wand führen (
61
,
166
), was auf eine unterdrückte BMPR-II-Signalgebung bei PAH zurückgeführt wurde (
166
). Aufgrund seiner Wirkung auf die SMC-Hyperplasie kann TNF auch die PA-Versteifung bei PH fördern; direkte Zusammenhänge zwischen TNF-Werten und PA-Versteifung bei PH müssen jedoch noch nachgewiesen werden. Bei anderen kardiovaskulären und entzündlichen Erkrankungen, z. B. Arteriosklerose, ist TNF ein etablierter Schlüsselmediator der Gefäßumgestaltung (
61
,
62
). Bei Patienten mit entzündlichen Arterienerkrankungen, nämlich rheumatoider Arthritis, ankylosierender Spondylitis und Psoriasis-Arthritis, die eine Anti-TNF-Behandlung mit Adalimumab, Etanercept oder Infliximab erhielten, wurde im Vergleich zu unbehandelten Kontrollpersonen eine Reduktion der Aortensteifigkeit gemessen anhand der Pulswellengeschwindigkeit und des Augmentationsindex festgestellt (
107
,
167
). Daher könnte die pharmakologische Hemmung von Entzündungsmediatoren wie IL-6 und TNF bei PH möglicherweise die Proliferation pulmonaler Gefäßzellen und die PA-Verdickung reduzieren und könnte daher eine gezielte Therapie für die PA-Versteifung darstellen.Darüber hinaus können proinflammatorische Mediatoren bei Herz-Kreislauf-Erkrankungen eine Gefäßversteifung durch eine erhöhte Produktion von ECM-Komponenten, nämlich fibrillären und nichtfibrillären Kollagenen und Fibronektin durch residente Gefäßzellen induzieren (
168
). Nach einem Herzinfarkt sowie bei ischämischer und nicht-ischämischer Herzinsuffizienz induzieren proinflammatorische Mediatoren wie TGF-β (
74
,
169
) und IL-1β (
170
) die Umwandlung von Fibroblasten in Myofibroblasten, die reichlich ECM-Proteine produzieren können (
168
) (
Tabellen 1
,
2
;
Abbildung 2
). Bei PH tragen adventitiale Myofibroblasten durch die Produktion struktureller ECM-Komponenten wie Kollagene, Elastin, Fibronektin und dynamischer ECM-Bestandteile wie Tenascin-C und Osteopontin (
43
,
46
,
74
) (
Tabelle 2
;
Abbildung 2
)
zur Umgestaltung und Versteifung der PA bei (46). Tenascin-C und Osteopontin wiederum erhöhen die Proliferation von Fibroblasten und SMC, was zur Umwandlung von Myofibroblasten und Mediaverdickung und damit zur Gefäßversteifung beiträgt (
43
,
46
,
171
) (
Abbildung 2
). In die pulmonale Adventitia rekrutierte aktivierte Makrophagen können ECM-Proteine wie Kollagen Typ I exprimieren und so zur ECM-Versteifung bei PH beitragen (
172
). In Tiermodellen mit MCT-induzierter PH wurde auch eine Hochregulierung der NADPH-Oxidase 4 (Nox4) in der pulmonalen Adventitia festgestellt, wo sie die TGF-β-vermittelte Expression von Matrixkollagenen durch Adventitiafibroblasten und damit die ECM-Versteifung fördert (
172
). In ähnlicher Weise wurde in einem Tiermodell mit chronischer hypoxischer PH eine erhöhte Kollagenablagerung durch residente Fibroblasten in der Adventitia festgestellt, was zu einer dickeren und steiferen Arterienwand führte (
172
–
174
). Um unlösliche, starre Fasern zu bilden, werden überschüssige fibrilläre Kollagene dann durch Vernetzungsenzyme weiter vernetzt (
43
,
175
). Insbesondere eine erhöhte Expression von LOX in SMCs und eine Expression des Lysyloxidase-ähnlichen Enzyms (LOXL) in Adventitiafibroblasten führt zu einer erhöhten Kollagenvernetzung und PA-Versteifung bei PAH (
176
). Darüber hinaus weisen Adventitiafibroblasten per se einen proinflammatorischen Phänotyp bei PH auf, einschließlich der Rekrutierung und Aktivierung von Adventitiamakrophagen (
7
) und zur Produktion proinflammatorischer Marker wie der Chemokine MCP-1, SDF-1, RANTES/CCR5, CCR7, CXCR4 und der Kostimulationsmoleküle CD40 und CD40L (
7
,
71
). Diese sekretorische Aktivität kann wiederum eine Rückkopplungsschleife erzeugen, die weitere Entzündungen und damit eine Umgestaltung der ECM auslöst.Neben erhöhten Konzentrationen zirkulierender Entzündungsmediatoren induziert erhöhter mPAP-Spiegel bei PH auch die Aktivierung des proinflammatorischen NF-κB-Signalwegs in PA ECs und SMCs (
58
,
133
,
157
) (
Abbildung 2
). Basierend auf Studien zu systemischen Herz-Kreislauf-Erkrankungen erweist sich eine solche Aktivierung von NF-κB als potenziell wichtiger Schritt bei der PA-Versteifung. So wurde gezeigt, dass nukleäres NF-κB die Expression von Aortenkollagen Typ I in einem Mausmodell für Typ-2-Diabetes erhöht, was zu einer Aortenversteifung führt, die ex vivo durch Druckmyographie gemessen wurde (
177
). Interessanterweise wurden diese Effekte durch eine NF-κB-abhängige Überexpression von RUNX2 vermittelt, einem wichtigen Transkriptionsfaktor, der nicht nur für die ECM-Umgestaltung [durch erhöhte Expression von ECM-Kollagenen durch SMCs (
177
)] relevant ist, sondern auch im Zusammenhang mit der Gefäßverkalkung (
55
,
177
) (wie unten erläutert) (
Abbildung 2
). Es lässt sich spekulieren, dass die Aktivierung von NF-κB ähnliche Effekte bei PH haben und so zur PA-Versteifung durch ECM-Umgestaltung und Gefäßverkalkung beitragen könnte.Der entzündungsbedingten Überproduktion von ECM-Komponenten bei Herz-Kreislauf-Erkrankungen steht ein erhöhter proteolytischer Abbau der ECM infolge eines parallelen Anstiegs von MMPs gegenüber (
46
). Bei PH sezernieren aktivierte Makrophagen und Myofibroblasten in der Adventitia MMPs, insbesondere MMP-2 (
154
,
162
), MMP-9 (
6
,
162
), MMP-10 (
47
) und MMP-19 (
6
,
154
), während Gewebeinhibitoren von Metalloproteinasen (TIMPs) herunterreguliert zu sein scheinen (
46
,
162
) (
Tabelle 2
;
Abbildung 2
). MMP-2 (
50
) und MMP-9 (
49
) bauen Elastin ab und verringern dadurch die Gefäßcompliance, was zu einer Arterienversteifung führt (
49
–
52
). Darüber hinaus erleichtert der Abbau elastischer Fasern und anderer ECM-Bestandteile wie BM-Kollagen, interstitieller Kollagene, Fibronektin und verschiedener Proteoglykane durch MMPs die Migration von Adventitiafibroblasten und Myofibroblasten in die Media und Intima, was wiederum die PA-Versteifung und Gefäßstenose fördert (
46
,
154
) (
Tabelle 2
). In ähnlicher Weise wird die Neointimalbildung durch erhöhte Proliferation und Migration von SMC aus der Media in die Intimabereiche der Arterienwand durch MMP-regulierten ECM-Abbau erleichtert (
52
,
178
) und fördert Gefäßstenose und -versteifung (
178
). Produkte der ECM-Proteolyse – die Matrikine [kürzlich ausführlich besprochen von Mutgan et al. (
179
)] – können wiederum als entzündungsfördernde Mediatoren dienen, die die Entzündung verstärken und so eine weitere positive Rückkopplungsschleife erzeugen können (
43
). Darüber hinaus ermöglicht der Abbau der ECM, dass zirkulierende Serumfaktoren in das Medium gelangen und die Produktion von Serinelastase durch SMCs stimulieren (
178
). Diese Serinelastasen unterstützen den Abbau von Elastin und die Freisetzung aktivierter Wachstumsfaktoren wie Fibroblastenwachstumsfaktor (FGF) und TGF-β, die wiederum die Produktion von Kollagen, Fibronektin und Tenascin-C durch SMCs und Fibroblasten erhöhen (
43
) – was wiederum die PA-Versteifung fördert (
Tabelle 2
). Bei anderen Herz-Kreislauf-Erkrankungen wie ischämischer Herzinsuffizienz sezernieren Immunzellen wie Makrophagen, Lymphozyten und Mastzellen MMPs, die die vaskuläre und kardiale ECM als Reaktion auf mechanische Belastung umgestalten (
168
). Bei Arteriosklerose waren erhöhte Konzentrationen von MMP-2 und MMP-9 mit einer erhöhten Arteriensteifigkeit und einem erhöhten Risiko für Herz-Kreislauf-Erkrankungen verbunden, was auf ihre Fähigkeit zurückgeführt wurde, die elastischen Schichten in den Arterien abzubauen (
180
,
181
). Dementsprechend reduziert der MMP-2-Knockdown die Arteriensteifigkeit der Halsschlagadern bei Mäusen, indem er den Elastinabbau im Gewebe verringert (
182
).So können sich die Aktivierung von Immunzellen und Entzündungswegen sowie die Verdickung der Arterienwände und die Umgestaltung der extrazellulären Matrix (ECM) gegenseitig stimulieren. Die gezielte Beeinflussung entzündlicher Prozesse bei Herz-Kreislauf-Erkrankungen, z. B. Aortenaneurysmen, hat sich als vorteilhaft für Schlüsselmechanismen der Umgestaltung der extrazellulären Matrix (ECM) erwiesen, wie z. B. Elastinabbau, MMP-Expression und Makrophageninfiltration (
183
). Ein besseres Verständnis der spezifischen Akteure und molekularen Wege, die an dieser gegenseitigen Interaktion beteiligt sind, kann neue und möglicherweise personalisierte Ziele für die zukünftige PH-Therapie aufzeigen.Verkalkung und Entzündung der LungenarterienBiologisch induzierte Mineralisierung ist ein integraler Bestandteil der menschlichen Physiologie und Gewebehomöostase. Dabei werden extra- und intrazelluläre Mechanismen eingesetzt, um die Bildung, das Wachstum und die Lokalisierung der abgelagerten Mineralien zu steuern. Unter Krankheitsbildern können diese Prozesse durch Veränderungen des lokalen oder globalen Kalziummilieus, DNA-Schäden, Stress des endoplasmatischen Retikulums, oxidativen Stress oder Stoffwechselstörungen – also Prozesse, die häufig mit Entzündungsreaktionen einhergehen – aus dem Gleichgewicht geraten und letztendlich zu pathologischer Verkalkung von Gewebe oder Blutgefäßen führen (
184
,
185
). Mechanistisch gesehen führen diese Faktoren zu (oder werden begleitet von) einer phänotypischen Umwandlung verschiedener Zelltypen in Osteoprogenitorzellen durch eine De-novo- bzw. erhöhte Expression des potenten Transkriptionsaktivators RUNX2, der die Expression nachgeschalteter, kalzifizierungsfördernder Proteine wie alkalischer Phosphatase auslöst (
186
–
188
). Im Vergleich zu systemischen Arterien wird die Gefäßverkalkung der PA kaum thematisiert, obwohl sie tatsächlich ein häufiges Merkmal bei Patienten mit schwerer, lang anhaltender PH (
189
), fortgeschrittener PH und PH mit chronischem Nierenversagen (
190
) oder terminaler Niereninsuffizienz (
191
) ist. Tatsächlich sagt der Nachweis einer Verkalkung der peripheren PA mittels Computertomographie (CT) (
192
) den Langzeitverlauf bei PH (
193
) und bei Patienten mit Vorhofseptumdefekt und Eisenmenger-Syndrom (
194
) voraus.Im Zusammenhang mit PAH wurde einer microRNA-204-abhängigen Hochregulierung von RUNX2 eine entscheidende Rolle bei der Regulierung der PA-Kalzifizierung zugeschrieben, die wiederum HIF-1α aktiviert, was zu einer Hyperproliferation von PA-SMCs, einer Resistenz gegen Apoptose und einer anschließenden Transdifferenzierung in osteoblastenähnliche Zellen führt (
55
). Eine zweite Studie berichtete, dass Hypoxie-induzierte zirkuläre RNA CDR1 die osteogene Transdifferenzierung menschlicher PA-SMCs fördert, indem sie microRNA-7-5p aufsaugt und infolgedessen ihre nachgeschalteten Zielmoleküle Calcium/Calmodulin-abhängige Kinase II-delta (CAMK2D) und Calponin 3 (CNN3) hochreguliert (
195
). Drittens steht die PA-Verkalkung in Zusammenhang mit Hypoxie, da Hypoxie die Expression der in den Granula von T-Lymphozyten und natürlichen Killerzellen gespeicherten Serinprotease Granzym B verringert, was die speichergesteuerten Calciumkanäle (SOCCs) als Hauptquelle des Minerals Calcium hemmt, indem es nicht-kanonische Wnt-Signale in SMCs abschwächt und so die Verkalkung der PA verstärkt (
141
). Unabhängig vom zugrundeliegenden Signalweg erhöht die Verkalkung letztlich die Gefäßsteifigkeit und verringert die Compliance der Pulmonalarterienwand, was ein Ausdruck der schlechten Prognose bei PH ist (
21
).Es wurde gezeigt, dass Entzündungssignale – wie sie bei PH auftreten – im systemischen Gefäßsystem vaskuläre Verkalkungsprozesse regulieren. Insbesondere fördert TNF die osteogene Differenzierung und Verkalkung boviner aortaler SMCs, indem es die Expression von Osteoblastenmarkern wie osteoblastenspezifischem Faktor 2 (Osf2), Aktivatorprotein 1 (AP1) und cAMP-responsivem Element-Bindungsprotein (CREB) durch Aktivierung der cAMP-Signalgebung induziert (
104
). Ebenso verursachte die Behandlung aortaler SMCs mit IL-1β oder IL-6 eine dosisabhängige Erhöhung der alkalischen Phosphataseaktivität und eine erhöhte Zellmineralisierung in vitro (
196
). Interessanterweise wird die Expression der inflammatorischen Zytokine IL-6, TNF und MCP-1 in verschiedenen Geweben epigenetisch durch das Bromodomain-Protein 4 (BRD4) reguliert (
86
), das die Chromatinlandschaft moduliert und die Genexpression aktiviert, indem es Transkriptionsfaktoren an Genpromotoren und/oder Superenhancern aufbaut. Insbesondere ist BRD4 in PA-SMCs von PAH-Patienten und in Lungen oder distalen PAs von Ratten-PH-Modellen hochreguliert und wird posttranskriptionell durch microRNA-204 reguliert (
87
), das gleichzeitig an der PA-Verkalkung beteiligt ist (
55
), wodurch eine zusätzliche epigenetische Verbindung zwischen Entzündung und Gefäßverkalkung hergestellt wird. Wichtiger noch: Es wurde gezeigt, dass der RUNX2-Genpromotor sowohl während der Osteoblastendifferenzierung (
197
) als auch bei Krebs (
198
) unter direkter Kontrolle von BRD4 steht, was nahelegt, dass BRD4 als „Hauptregulator“ sowohl der Entzündung als auch der Gefäßverkalkung gleichzeitig dienen kann. In Übereinstimmung mit dieser Annahme schwächte die BRD4-Hemmung die Umgestaltung der Lungen- und Koronararterien bei experimenteller PH ab, und dieser Schutzeffekt war mit verringerten IL-6- und MCP-1-Spiegeln verbunden (
199
,
200
).Obwohl es nur wenige Studien gibt, die Verkalkung und Entzündung bei PH in Zusammenhang bringen, sind Zytokine an der Regulierung der Verkalkung in den extrapulmonalen Gefäßen beteiligt. Wichtig ist, dass die Gefäßverkalkung auch eng mit der Umgestaltung und Versteifung der ECM verknüpft zu sein scheint (
201
), da die Mineralisierung der SMC direkt mit der Produktion von Kollagen I und dem Abbau von Fibronektin und Elastin korreliert, während letzteres Gerüste für die Kalziumeinlagerung bildet (
201
–
203
). Diese Erkenntnisse legen nahe, dass vorgelagerte Entzündungen durch die Umgestaltung der ECM auch die Gefäßverkalkung fördern können.Pulmonalarterieller endothelialer-mesenchymaler Übergang und EntzündungWährend sich unsere Untersuchung von Gefäßverkalkungsprozessen weitgehend auf SMCs konzentriert hat, ist es wichtig zu bedenken, dass auch ECs beteiligt sind. Bei verschiedenen Herz-Kreislauf-Erkrankungen verlieren ECs ihre charakteristische Morphologie und verändern sich hin zu einem mesenchymalen Phänotyp (
204
), ein Prozess, der als endothelial-mesenchymaler Übergang (EndoMT) bezeichnet wird und insbesondere durch Entzündungen moduliert wird. Insbesondere wurde gezeigt, dass entzündliche Zytokine wie IL-1β oder TNF EndoMT in PA ECs induzieren. Im Gegenzug beginnen diese EndoMT-Zellen, entzündliche Zytokine wie IL-4, IL-6, IL-8, IL-13 und TNF in viel höheren Konzentrationen als normale PA ECs abzusondern (
205
), wodurch eine potenziell bösartige Feedforward-Schleife entsteht. Im Einklang mit der Theorie der entzündungsbedingten EndoMT bei PH zeigte sich, dass die Aktivierung des proinflammatorischen NF-κB-Signalwegs in einem Mausmodell der durch Monocrotalin (MCT) induzierten PH zu einer Hochregulierung von miR-130a führte, was zu einem Verlust des Knochenmorphogenetischen Proteinrezeptors Typ 2 (BMPR2), einer erhöhten Expression von HMGA1 (High Mobility Group AT-hook 1) und schließlich zu EndoMT in mikrovaskulären endothelialen Zellen der Lunge führte (
206
). Es ist wichtig hervorzuheben, dass EndoMT zwar ausführlich in pulmonalen und systemischen endokrinen Zellen (ECs), die Entzündungsmediatoren ausgesetzt waren, in vitro dokumentiert wurde , das Ausmaß und die Relevanz von EndoMT in vivo in jüngsten Studien unter Verwendung von Technologien zur Abstammungsverfolgung jedoch weiterhin umstritten sind: Durch Verwendung von doppelt transgenen Mäusen, die in allen ECs stabil grünes fluoreszierendes Protein (GFP) exprimieren, konnten Suzuki und Kollegen GFP in 14,3 ± 1,8 % der mesenchymalen (CD144 - CD45 - CD326 - ) Zellen nachweisen, was auf eine substantielle EndoMT hinweist (
207
). In ähnlicher Weise zeigte die Endothel-Linienverfolgung unter Verwendung transgener vaskulärer Endothel-Cadherin-Cre-Rekombinase oder Tie-2 Cre-Mäuse, die mit mTomato/mGreen Fluoreszenzprotein-Doppelfluoreszenz-Cre-Reportermäusen gekreuzt wurden, reichlich endotheliale Linien-markierte Zellen in der Neointima, wo sie nach Induktion von PH durch Monocrotalin-Pyrrol glatte Muskel-α-Aktin und glatte Muskel-Myosin-Schwerketten exprimierten (
208
). Eine kürzlich durchgeführte Linienverfolgungsstudie in chronischen Hypoxie- und allergeninduzierten Modellen der Lungengefäßumgestaltung zeigte jedoch die Beibehaltung endothelialer Linien-spezifischer Marker-Expressionsprofile ohne Anzeichen einer Zelltypumwandlung (
209
). Insbesondere steht die Erkennung einer eingeschränkten oder partiellen EndoMT nicht unbedingt im Widerspruch zu ihrer potenziellen funktionellen Relevanz bei der PA-Versteifung, sondern legt lediglich nahe, dass diese Relevanz möglicherweise eher mit der Freisetzung proliferativer, hypertropher und profibrotischer Signale – also Mediatoren von Prozessen, die letztlich die PA-Versteifung fördern – durch partielle EndoMT-Zellen zusammenhängt als mit der tatsächlichen Bildung einer signifikanten mesenchymalen Zellmasse über diesen Mechanismus. Tatsächlich wird eine ähnliche Rolle zunehmend für den epithelial-mesenchymalen Übergang bei Gewebefibrose erkannt (
210
).Darüber hinaus kann EndoMT Entzündungen mit Gefäßverkalkung und damit mit PA-Versteifung bei PH in Verbindung bringen. Insbesondere Studien an aortischen endothelialen Zellen (ECs) zeigen, dass inflammatorische Zytokine wie TNF und IL-1β EndoMT modulieren und die Expression der BMPR2- und JNK-Signalgebung herunterregulieren, wodurch ECs für BMP9-induzierte osteogene Differenzierung sensibilisiert werden, die in Mineralisierung gipfelt (
141
). Ähnliche Regulationsmechanismen können die Verkalkung von PA-ECs bei verschiedenen Arten von PH vorantreiben, und PAH-Patienten mit BMPR2-Mutationen oder Beeinträchtigungen des BMP-Signalwegs (
104
) wären in diesem Szenario voraussichtlich besonders anfällig, da eine Beeinträchtigung der BMPR2-Signalgebung mit EndoMT in Zusammenhang steht (
211
). Abstammungsverfolgungsstudien im systemischen Kreislauf unterstützen eine Rolle von EndoMT bei der Gefäßverkalkung. Sie zeigen beispielsweise, dass eine Untergruppe von Endokardzellen eine endokardial-mesenchymale Transition durchlaufen kann, die zur Verkalkung der Herzklappen von Mäusen und Menschen führt (
212
), oder dass vaskuläre endokardiale Zellen in osteogene Zellen übergehen können (
213
), was durch Hemmung der Glykogensynthase-Kinase 3 (GSK3) verhindert werden kann (
214
). Die Rolle von EndoMT (oder partiellem EndoMT) bei der Gefäßverkalkung im Lungenkreislauf und im Zusammenhang mit PH wurde bisher jedoch nicht untersucht.Mögliche klinische RelevanzWährend sich aktuelle PH-Therapien (z. B. Prostazykline, Phosphodiesterasehemmer, Kalziumkanalblocker, Endothelin-Rezeptorantagonisten oder Stimulatoren der löslichen Guanylatcyclase) in erster Linie auf die Linderung der Vasokonstriktion als symptomatischen Ansatz konzentrieren (
215
), besteht das langfristige therapeutische Ziel darin, sich auf die Mechanismen zu konzentrieren, die für den Ausbruch und das Fortschreiten der Krankheit verantwortlich sind, darunter Gefäßumbau und Entzündung (
215
). In dieser Hinsicht könnte das gezielte Angreifen der Immun-PA-Versteifungsachse eine besonders vielversprechende Strategie sein, angesichts der prädiktiven und pathomechanistischen Rolle der PA-Versteifung bei PH und des Arsenals immunmodulatorischer Therapien, die sich bereits im klinischen Einsatz oder in der Entwicklung befinden. Im systemischen Kreislauf haben sich entzündungshemmende Therapien als vielversprechend bei der Verringerung der Arterienversteifung bei entzündlichen Arterienerkrankungen wie rheumatoider Arthritis oder Psoriasis-Arthritis erwiesen (
167
). Insbesondere TNF-Antagonisten wie Adalimumab, Etanercept oder Infliximab sind etablierte entzündungshemmende Therapien bei (Auto-)Immunerkrankungen (
216
), die die Aortensteifigkeit
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.