
- Beiträge: 1757
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Glatte Muskulatur Säureempfindlicher Ionenkanal 1a als therapeutisches Ziel ...
Glatte Muskulatur Säureempfindlicher Ionenkanal 1a als therapeutisches Ziel ...
07 Jul 2024 14:16 #2150
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Glatte Muskulatur Säureempfindlicher Ionenkanal 1a als therapeutisches Ziel ... wurde erstellt von danny
Glatte Muskulatur Säureempfindlicher Ionenkanal 1a als therapeutisches Ziel zur Umkehrung der hypoxischen pulmonalen Hypertonie
www.frontiersin.org/journals/molecular-b...olb.2022.989809/full
Der säureempfindliche Ionenkanal 1a (ASIC1a) ist ein spannungsunabhängiger, nicht selektiver Kationenkanal, der sowohl Na + als auch Ca2 + leitet . Die Aktivierung von ASIC1a löst eine Depolarisation der Plasmamembran aus und stimuliert intrazelluläre Ca2 + -abhängige Signalwege in mehreren Zelltypen, darunter Gefäßglattmuskelzellen (SM) und Endothelzellen (ECs). Frühere Studien haben gezeigt, dass ein Anstieg des pulmonalen Gefäßwiderstands, der mit chronischer Hypoxie (CH) induzierter pulmonaler Hypertonie einhergeht, ASIC1a erfordert, um eine verstärkte pulmonale Vasokonstriktion und Gefäßumgestaltung hervorzurufen. Sowohl SM- als auch EC-Dysfunktion treiben diese Prozesse an; die Beteiligung von ASIC1a an diesen verschiedenen Zelltypen ist jedoch unbekannt. Mithilfe des Cre-LoxP-Systems zur Erzeugung zelltypspezifischer Asic1a -Knockout-Mäuse testeten wir die Hypothese, dass SM- Asic1a zu CH-induzierter pulmonaler Hypertonie und Gefäßumbau beiträgt, während EC- Asic1a der Entwicklung von CH-induzierter pulmonaler Hypertonie entgegenwirkt. Der Schweregrad der pulmonalen Hypertonie blieb bei Mäusen mit spezifischer Deletion von EC- Asic1a (Tek Cre - Asic1a fl/fl ) unverändert. Ähnlich wie globale Asic1a- Knockout-Mäuse ( Asic1a −/- ) waren Mäuse mit spezifischer Deletion von SM- Asic1a (MHC CreER - Asic1a fl/fl ) jedoch vor der Entwicklung von CH-induzierter pulmonaler Hypertonie und Rechtsherzhypertrophie geschützt. Darüber hinaus war die pulmonale Hypertonie rückgängig gemacht worden, als die Deletion von SM- Asic1a bei konditionellen MHC CreER - Asic1a fl/fl- Mäusen mit bestehender pulmonaler Hypertonie eingeleitet wurde. Die durch CH induzierte Gefäßumgestaltung war auch in den Lungenarterien von MHC CreER - Asic1a fl/fl- Mäusen deutlich abgeschwächt. Diese Ergebnisse wurden zusätzlich durch eine verringerte, durch CH induzierte Proliferation und Migration von pulmonalarteriellen glatten Muskelzellen (PASMCs) von Asic1a −/- Mäusen unterstützt. Zusammen zeigen diese Daten, dass SM-, aber nicht EC- Asic1a zu durch CH induzierter pulmonaler Hypertonie und Gefäßumgestaltung beiträgt. Darüber hinaus liefern diese Studien Beweise für das therapeutische Potenzial der ASIC1a-Hemmung zur Umkehrung der pulmonalen Hypertonie. 1. EinleitungUnter normalen physiologischen Bedingungen wird der Lungenkreislauf in einem Zustand mit niedrigem Druck und geringem Widerstand gehalten, mit geringem oder keinem Ruhegefäßtonus. Unter pathologischen Bedingungen führt ein anhaltender Anstieg des pulmonalen Gefäßwiderstands zur Entwicklung einer pulmonalen Hypertonie. Pulmonale Hypertonie ist eine fortschreitende und häufig tödlich verlaufende Lungengefäßerkrankung, die durch einen mittleren pulmonalarteriellen Druck von >20 mmHg definiert ist ( Simonneau et al., 2019 ). Mit der Zeit erhöhen der erhöhte Gefäßwiderstand und der pulmonalarterielle Druck die Nachlast des rechten Ventrikels. Wenn die Anpassungsmechanismen der Dilatation und Hypertrophie des rechten Ventrikels den hohen Gefäßwiderstand in der Lunge nicht mehr kompensieren können, tritt eine Rechtsherzinsuffizienz auf, die mit einer schlechten Prognose verbunden ist.Obwohl pulmonale Hypertonie verschiedene Ursachen haben kann, kann der Anstieg des pulmonalen Gefäßwiderstands bei allen Formen der pulmonalen Hypertonie auf eine Kombination aus anhaltender pulmonaler Vasokonstriktion und Gefäßumbau zurückgeführt werden. Eine verstärkte Vasokonstriktion ist mit einer Funktionsstörung der pulmonalarteriellen Endothelzellen (PAEC) und einer Hyperreaktivität der pulmonalarteriellen glatten Muskelzellen (PASMCs) verbunden ( Budhiraja et al., 2004 ; Lin et al., 2004 ; Nagaoka et al., 2004 ; Jernigan et al., 2008 ; Broughton et al., 2010 ; Weise-Cross et al., 2018 ). Die Auswirkungen der Umgestaltung auf den pulmonalvaskulären Widerstand sind vor allem auf die Verdickung der Intima und/oder Mediaschicht kleiner Muskelarterien und die distale Neomuskularisierung zurückzuführen, die sich durch das Auftreten von Zellen äußert, die glatte Muskelzellen (SM)-spezifische Marker in normalerweise nicht-muskulären präkapillären, intraazinären Gefäßen exprimieren. Diese komplexe Pathogenese wird vermutlich durch eine Verletzung und Apoptose von Endothelzellen (EC) eingeleitet, gefolgt von der Entstehung übermäßiger Proliferation und Migration apoptoseresistenter PAECs und PASMCs sowie zellulärer Transdifferenzierung in Form eines EC-mesenchymalen Übergangs und phänotypischer SM-Transformationen ( Voelkel & Tuder, 2000 ; Shimoda & Laurie, 2013 ; Gao et al., 2016 ). Stoffwechselstörungen, die die aerobe Glykolyse fördern und die oxidative Atmung der Mitochondrien hemmen, führen nachweislich sowohl bei Tiermodellen als auch bei Patienten mit pulmonaler Hypertonie zu umfassenden Umbauten des rechten Ventrikels und der Gefäße ( McMurtry et al., 2004 ; Bonnet et al., 2006 ; Xu et al., 2007 ; Sutendra et al., 2010 ; Fessel et al., 2012 ; Dromparis et al., 2013 ; Pak et al., 2013 ). Die Umstellung des Zellstoffwechsels auf Milchsäuregärung führt zu einem pathologischen Anstieg des extrazellulären Säuregehalts. Mehrere Ionenkanäle werden entweder direkt gesteuert oder ihre Aktivität wird durch Änderungen des intra- und extrazellulären pH-Werts moduliert. Dazu gehören die säureempfindlichen Ionenkanäle (ASIC), der transiente Rezeptorpotential-Vanilloid-Rezeptor 1 (TRPV1), der transiente Rezeptorpotential-Ankyrin-Repeat-Rezeptor 1 (TRPA1), einige Zwei-Poren-Domänen-Kanäle (K2P), nach innen gleichrichtende K + -Kanäle (Kir) sowie spannungsgesteuerte Na +- , Ca2 + - und K + -Kanäle ( Harguindey et al., 2017 ).Säureempfindliche Ionenkanäle (ASICs) stellen eine Unterfamilie der Amilorid-sensitiven Degenerin/Epithel-Na + -Kanäle (Deg/ENaC) dar, die H + -gesteuerte, spannungsunempfindliche Kationenkanäle bilden. Ähnlich wie ENaCs sind ASICs hochselektiv für Na + gegenüber anderen Ionen, mit Ausnahme von ASIC1a, das zusätzlich Ca2 + leitet ( Waldmann et al., 1997 ; Xiong et al., 2004 ; Yermolaieva et al., 2004 ). Der Einstrom von Na + und Ca2 + trägt zur Membrandepolarisation, Aktivierung von Ca2 + -Calmodulin-abhängigen Mechanismen und anderen Second-Messenger-Wegen bei, was die vielfältigen Rollen verdeutlicht, die ASIC1a bei der intrazellulären Signalgebung und Erregbarkeit unter normalen wie auch pathologischen Bedingungen spielt. ASICs wurden vor allem in Neuronen untersucht, da sie im gesamten zentralen und peripheren Nervensystem ubiquitär exprimiert werden. Folglich ist es weniger bekannt, dass ASICs in einer Vielzahl anderer Zelltypen exprimiert werden, darunter Oligodendrozyten, Mesenchym-, Epithel-, Endothel-, Muskel-, Fett-/Endokrin- und Immunzellen, wo sie mit einer Reihe von Pathologien in Verbindung gebracht wurden ( Foster et al., 2021 ; Karlsson et al., 2021 ). Obwohl die Expression von ASIC1 in vaskulären SM und ECs berichtet wurde ( Grifoni et al., 2008 ; Jernigan et al., 2009 ; Chung et al., 2010 ; Akanji et al., 2019 ; Garcia et al., 2020 ; Redd et al., 2021 ), ist über die funktionelle Rolle von ASIC1 bei der Regulierung der vaskulären Homöostase bei Krankheitszuständen weniger bekannt.Frühere Studien in unserem Labor haben eine neue Rolle von ASIC1a bei der Entwicklung einer durch chronische Hypoxie (CH) induzierten pulmonalen Hypertonie identifiziert ( Nitta et al., 2014 ). ASIC1 wird sowohl in PASMCs als auch in PAECs exprimiert. Der Beitrag von ASIC1a zu den pathologischen Mechanismen, die zur Umgestaltung der Pulmonalarterien und zur Entwicklung von pulmonaler Hypertonie in diesen beiden Gefäßzelltypen führen, ist jedoch unklar. Während frühere Studien darauf hinweisen, dass PASMC ASIC1a eine pulmonale Vasokonstriktion vermittelt ( Jernigan et al., 2012 ), ist die funktionelle Rolle von ASIC1 in PAECs unbekannt. Basierend auf Studien, die zeigen, dass EC ASIC1 in Mesenterialarterien zur endothelialabhängigen Vasodilatation beiträgt ( Garcia et al., 2018 ), spekulieren wir, dass PAEC ASIC1a vor der Entwicklung von pulmonaler Hypertonie schützen könnte. Um die Hypothesen zu testen, dass SM- Asic1a zu CH-induzierter pulmonaler Hypertonie und vaskulärer Umgestaltung beiträgt und EC- Asic1a der Entwicklung von CH-induzierter pulmonaler Hypertonie entgegenwirkt, werden wir das Cre-loxP-System verwenden, um Mäuse mit selektiver EC- Asic1a- Deletion (Tek Cre - Asic1a fl/fl ) oder induzierbarer SM- Asic1a- Deletion (MHC CreER - Asic1a fl/fl ) zu erzeugen.Materialen und MethodenEthische GenehmigungAlle in dieser Studie verwendeten Protokolle wurden vom Institutional Animal Care and Use Committee der University of New Mexico School of Medicine (Protokoll Nr. 19-200899-HSC) geprüft und genehmigt und entsprechen den Richtlinien der National Institutes of Health für den Umgang mit Tieren. Alle Tiere wurden mit einer Überdosis Pentobarbital-Natrium (200 mg/kg, ip) betäubt und nach dem Bewusstseinsverlust sofort durch Ausbluten getötet.TiereDie Studien wurden an erwachsenen männlichen Wildtyp-Mäusen ( Asic1a +/+ ) oder verschiedenen transgenen Mäusen (12–16 Wochen alt) durchgeführt, wie in Tabelle 1 gezeigt. Um Asic1a in ECs oder SM selektiv zu löschen , wurden Asic1a fl/fl -Mäuse mit Tek Cre- bzw. MHC CreER -transgenen Mäusen gekreuzt. Es wurden homozygote und/oder heterozygote Mäuse gezüchtet und die Cre-Transgenexpression sowie die Deletion des Asic1a -Gens wurden durch PCR und Agarosegelelektrophorese bestätigt ( Tabelle 1 ). Die Tiere wurden eins bis fünf pro Käfig in einer speziellen pathogenfreien (SPF) Tierpflegeeinrichtung untergebracht und bei einem Hell-Dunkel-Rhythmus von 12:12 Stunden gehalten. Standardfutter (Teklad sojaproteinfreie Diät Nr. 2920, Envigo) und Wasser wurden ad libitum bereitgestellt . Die Tiere wurden zufällig den Versuchsgruppen zugeteilt und die Genotyp- und Behandlungszuweisungen wurden den Forschern wenn möglich nicht mitgeteilt. Es wurden ausschließlich männliche Mäuse untersucht, da die Expression von iCreER T2 unter der Kontrolle des SM-Promoters auf dem Y-Chromosom eingefügt ist. Darüber hinaus zeigten unsere bisherigen Daten keine signifikante Interaktion zwischen Geschlecht und der Entwicklung von hypoxischer pulmonaler Hypertonie ( Nitta et al., 2014 ). Zur Induktion der Cre-Aktivität wurden MHC CreER - Asic1a fl/fl -Mäusen an fünf aufeinanderfolgenden Tagen einmal täglich 75 mg/kg Tamoxifen (TAM; Sigma-Aldrich, CAS-Nr. 10540-29–1) in Maisöl injiziert. Nach 14 Tagen wurde die Cre-Rekombinase anhand des Schwanz-DNA-Primerpaars 5'-LoxP-Vorwärts- und 3'-LoxP-Rückwärtsprimerpaars bestimmt ( Tabelle 1 ). Tamoxifen wurde MHC CreER - Asic1a fl/fl -Mäusen verabreicht , um einen SM- Asic1a- Knockout zu induzieren, entweder 1) 2 Wochen vor CH als vorbeugendes (pTAM) Protokoll oder 2) nach 3 Wochen CH als therapeutisches (tTAM) Protokoll, um die Umkehrung einer bestehenden pulmonalen Hypertonie zu beurteilen. Wir haben zuvor gezeigt, dass Mäuse nach 3-wöchiger CH-Exposition pulmonale Hypertonie entwickeln ( Sheak et al., 2020 ). Die Asic1a- Disruptierung wurde anhand der Gesamt-RNA (1 µg) im Gehirn (positive Kontrolle) und isolierten Lungenarterien mittels RT-PCR beurteilt. Die Gesamt-RNA wurde mit TRIzol extrahiert und in cDNA rücktranskribiert (Transcription First-Strand cDNA Synthesis Kit, Roche, 04379012001). Die Amplifikation von Asic1a erfolgte mittels PCR (iCycler, Bio-Rad) unter Verwendung von REDExtract-N_Amp PCR Ready Mix (Sigma-Aldrich, XNAT) und Asic1Primer: vorwärts: 5′ CACATGCCAGGGGATGCCCC 3′ und rückwärts: 5′ AGCCGGTGCTTAATGACCTC 3‘ (410 bp). Das PCR-Produkt wurde mittels Gelelektrophorese auf einem 3%igen Agarosegel aufgetrennt und zur Visualisierung unter UV-Licht mit Ethidiumbromid gefärbt. Tabelle 1
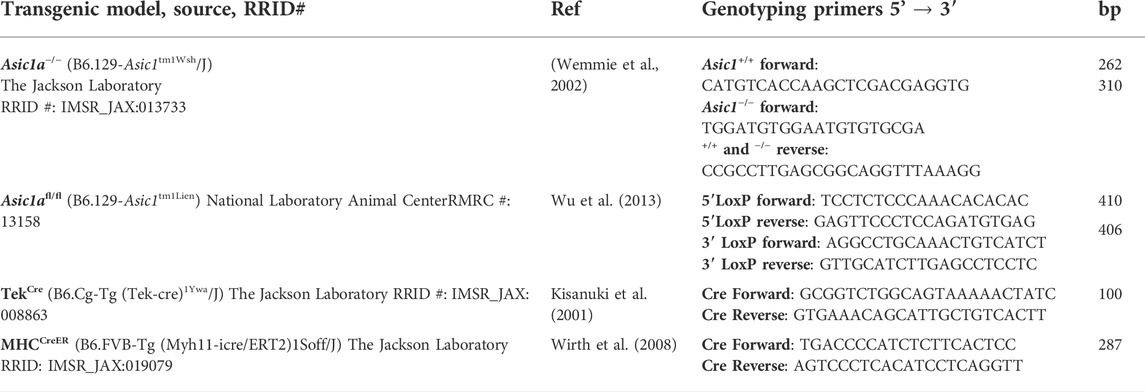 TABELLE 1. Transgene Mausmodelle, Quelle, Referenz, Primer und erwartete Basenpaare zur Identifizierung jedes Genotyps. Beurteilung des systemischen mittleren arteriellen BlutdrucksBlutdruck und Herzfrequenz wurden bei Mäusen mithilfe von Radiotelemetriegeräten (PA-C10-Implantat; Data Systems International) aufgezeichnet. Telemetriesender wurden unter sterilen Bedingungen mit inhalierter Isofluran-Anästhesie (2 % Isofluran und 98 % O 2 -Gasgemisch) chirurgisch implantiert. Das Analgetikum Buprenex (Buprenorphin; 0,1 mg/kg, IM) wurde vor Beginn der Operation verabreicht, um eine effektive Genesung und präventive Schmerzbehandlung zu gewährleisten. Unter Verwendung steriler Techniken wurde ein Mittellinienschnitt vorgenommen, um die Halsschlagader durch stumpfe Dissektion freizulegen. In der Halsschlagader wurde zwischen zwei Seidennähten ein kleiner Einschnitt vorgenommen und das Ende des Katheters einer kleinen implantierbaren PA-C10-Telemetriesonde eingeführt und in Richtung Herz vorgeschoben. Die Spitze wurde festgebunden und der Körper des Telemeters subkutan im Mittelflankenbereich der Halsschlagader befestigt. Die Wunde wurde mit sterilen Nähten geschlossen und die Maus konnte sich 5 Tage lang erholen, bevor der Blutdruck gemessen wurde. Der Blutdruck wurde 72 Stunden lang aufgezeichnet (alle 15 Minuten in 10-Sekunden-Intervallen) und die Daten wurden als 24-Stunden-Durchschnittswerte dargestellt.Exposition gegenüber CHCH ist eine häufige Komplikation chronischer Lungenerkrankungen und ein wichtiger Auslöser für die Entwicklung von pulmonaler Hypertonie. Tiere, die CH ausgesetzt werden sollten, wurden 6 Wochen lang in einer hypobaren Kammer aus klarem Plexiglas (∼0,5 m3) mit einem Luftdruck von ∼380 mmHg untergebracht . Die hypobare Kammer wurde mithilfe einer Vakuumpumpe teilweise evakuiert, sodass ein kontinuierlicher Luftstrom von 30 l/min durch die Kammer strömen konnte. Die Kammer wurde 2 Mal pro Woche geöffnet, um die Einstreu zu wechseln und frisches Wasser und Futter bereitzustellen. Altersgleiche Kontrolltiere wurden bei Umgebungsluftdruck (∼630 mmHg in Albuquerque, NM) untergebracht. Wir haben zuvor gezeigt, dass dieses Mausmodell viele der kardiopulmonalen Veränderungen nachahmt, die bei pulmonaler Hypertonie beim Menschen beobachtet werden, darunter erhöhter systolischer Druck im rechten Ventrikel, rechtsventrikuläre Hypertrophie, verstärkte Vasokonstriktion und arterielle Umgestaltung (
Nitta et al., 2014
;
Detweiler et al., 2019
;
Sheak et al., 2020
).Immunfluoreszenz aus paraffineingebettetem LungengewebeDie Mäuse wurden mit Natriumpentobarbital (200 mg/kg ip) anästhesiert. Nach einer medianen Sternotomie wurde Heparin (100 U/20 g Körpergewicht) direkt in den rechten Ventrikel injiziert und die Lungenarterie mit einer 22-Gauge-Zufuhrnadel kanüliert. Das Präparat wurde sofort mit 0,1 M PBS mit 10 –4 M Papaverin perfundiert, um die Gefäße maximal zu erweitern und den Blutkreislauf freizusetzen. Die Lungen wurden dann mit 25 ml Fixiermittel (0,1 M PBS mit 4 % Saccharose, 4 % Paraformaldehyd und 10 –4 M Papaverin) bei einem Druck von 50 cm H 2 O über dem Hilus perfundiert und die Trachea auf einen Druck von 25 cm H 2 O aufgepumpt, wodurch während der Fixierung ein transmuraler Dehnungsdruck von 25 cm H 2 O entstand, um sicherzustellen, dass die Gefäße vollständig erweitert wurden. Die Luftröhre wurde mit 4–0-Seide abgebunden und die Lungen über Nacht in ein Fixiermittel getaucht, dehydriert und dann in Paraffin montiert.Es wurden Abschnitte (5 μm dick) geschnitten und auf Superfrost Plus-Objektträger (Fisher Scientific) montiert. Die Antikörper-Antigen-Bindung wurde durch hitzevermittelte Antigen-Retrievalierung verstärkt, entweder mit Tris-EDTA-Puffer (10 mM Tris, 1 mM EDTA, 0,05 % Tween-20, pH 9) für 15 Min. bei 100 °C (für Ki-67) oder Zitronensäure-Natriumcitrat-Puffer (pH 6, 0,05 % Tween-20) für 25 Min. bei 100 °C (für Remodellierung und ASIC1-Expression). Die Abschnitte wurden mit primären (24 h bei 4 °C) und sekundären Antikörpern (24 h bei 4 °C) inkubiert, wie in
Tabelle 2
angegeben . Wir haben zuvor die Spezifität von Ziegen-Anti-ASIC1 anhand von Wildtyp- und Knockout-Mäusen bestimmt (
Nitta et al., 2014
). Die Abschnitte wurden mit FluoroGel (Electron Microscopy Sciences) montiert und Querschnittsbilder der Lungenarteriolen (<100 µm) wurden sequenziell durch konfokale Mikroskopie (TCS SP5, Leica) unter Verwendung von Argon- (488 nm/∼20 mW), HeNe- (543 nm/∼1 mW) und HeNe-Lasern (633 nm/∼10 mW) Klasse IIIb und einem ×63-Objektiv aufgenommen. Tabelle 2
TABELLE 1. Transgene Mausmodelle, Quelle, Referenz, Primer und erwartete Basenpaare zur Identifizierung jedes Genotyps. Beurteilung des systemischen mittleren arteriellen BlutdrucksBlutdruck und Herzfrequenz wurden bei Mäusen mithilfe von Radiotelemetriegeräten (PA-C10-Implantat; Data Systems International) aufgezeichnet. Telemetriesender wurden unter sterilen Bedingungen mit inhalierter Isofluran-Anästhesie (2 % Isofluran und 98 % O 2 -Gasgemisch) chirurgisch implantiert. Das Analgetikum Buprenex (Buprenorphin; 0,1 mg/kg, IM) wurde vor Beginn der Operation verabreicht, um eine effektive Genesung und präventive Schmerzbehandlung zu gewährleisten. Unter Verwendung steriler Techniken wurde ein Mittellinienschnitt vorgenommen, um die Halsschlagader durch stumpfe Dissektion freizulegen. In der Halsschlagader wurde zwischen zwei Seidennähten ein kleiner Einschnitt vorgenommen und das Ende des Katheters einer kleinen implantierbaren PA-C10-Telemetriesonde eingeführt und in Richtung Herz vorgeschoben. Die Spitze wurde festgebunden und der Körper des Telemeters subkutan im Mittelflankenbereich der Halsschlagader befestigt. Die Wunde wurde mit sterilen Nähten geschlossen und die Maus konnte sich 5 Tage lang erholen, bevor der Blutdruck gemessen wurde. Der Blutdruck wurde 72 Stunden lang aufgezeichnet (alle 15 Minuten in 10-Sekunden-Intervallen) und die Daten wurden als 24-Stunden-Durchschnittswerte dargestellt.Exposition gegenüber CHCH ist eine häufige Komplikation chronischer Lungenerkrankungen und ein wichtiger Auslöser für die Entwicklung von pulmonaler Hypertonie. Tiere, die CH ausgesetzt werden sollten, wurden 6 Wochen lang in einer hypobaren Kammer aus klarem Plexiglas (∼0,5 m3) mit einem Luftdruck von ∼380 mmHg untergebracht . Die hypobare Kammer wurde mithilfe einer Vakuumpumpe teilweise evakuiert, sodass ein kontinuierlicher Luftstrom von 30 l/min durch die Kammer strömen konnte. Die Kammer wurde 2 Mal pro Woche geöffnet, um die Einstreu zu wechseln und frisches Wasser und Futter bereitzustellen. Altersgleiche Kontrolltiere wurden bei Umgebungsluftdruck (∼630 mmHg in Albuquerque, NM) untergebracht. Wir haben zuvor gezeigt, dass dieses Mausmodell viele der kardiopulmonalen Veränderungen nachahmt, die bei pulmonaler Hypertonie beim Menschen beobachtet werden, darunter erhöhter systolischer Druck im rechten Ventrikel, rechtsventrikuläre Hypertrophie, verstärkte Vasokonstriktion und arterielle Umgestaltung (
Nitta et al., 2014
;
Detweiler et al., 2019
;
Sheak et al., 2020
).Immunfluoreszenz aus paraffineingebettetem LungengewebeDie Mäuse wurden mit Natriumpentobarbital (200 mg/kg ip) anästhesiert. Nach einer medianen Sternotomie wurde Heparin (100 U/20 g Körpergewicht) direkt in den rechten Ventrikel injiziert und die Lungenarterie mit einer 22-Gauge-Zufuhrnadel kanüliert. Das Präparat wurde sofort mit 0,1 M PBS mit 10 –4 M Papaverin perfundiert, um die Gefäße maximal zu erweitern und den Blutkreislauf freizusetzen. Die Lungen wurden dann mit 25 ml Fixiermittel (0,1 M PBS mit 4 % Saccharose, 4 % Paraformaldehyd und 10 –4 M Papaverin) bei einem Druck von 50 cm H 2 O über dem Hilus perfundiert und die Trachea auf einen Druck von 25 cm H 2 O aufgepumpt, wodurch während der Fixierung ein transmuraler Dehnungsdruck von 25 cm H 2 O entstand, um sicherzustellen, dass die Gefäße vollständig erweitert wurden. Die Luftröhre wurde mit 4–0-Seide abgebunden und die Lungen über Nacht in ein Fixiermittel getaucht, dehydriert und dann in Paraffin montiert.Es wurden Abschnitte (5 μm dick) geschnitten und auf Superfrost Plus-Objektträger (Fisher Scientific) montiert. Die Antikörper-Antigen-Bindung wurde durch hitzevermittelte Antigen-Retrievalierung verstärkt, entweder mit Tris-EDTA-Puffer (10 mM Tris, 1 mM EDTA, 0,05 % Tween-20, pH 9) für 15 Min. bei 100 °C (für Ki-67) oder Zitronensäure-Natriumcitrat-Puffer (pH 6, 0,05 % Tween-20) für 25 Min. bei 100 °C (für Remodellierung und ASIC1-Expression). Die Abschnitte wurden mit primären (24 h bei 4 °C) und sekundären Antikörpern (24 h bei 4 °C) inkubiert, wie in
Tabelle 2
angegeben . Wir haben zuvor die Spezifität von Ziegen-Anti-ASIC1 anhand von Wildtyp- und Knockout-Mäusen bestimmt (
Nitta et al., 2014
). Die Abschnitte wurden mit FluoroGel (Electron Microscopy Sciences) montiert und Querschnittsbilder der Lungenarteriolen (<100 µm) wurden sequenziell durch konfokale Mikroskopie (TCS SP5, Leica) unter Verwendung von Argon- (488 nm/∼20 mW), HeNe- (543 nm/∼1 mW) und HeNe-Lasern (633 nm/∼10 mW) Klasse IIIb und einem ×63-Objektiv aufgenommen. Tabelle 2
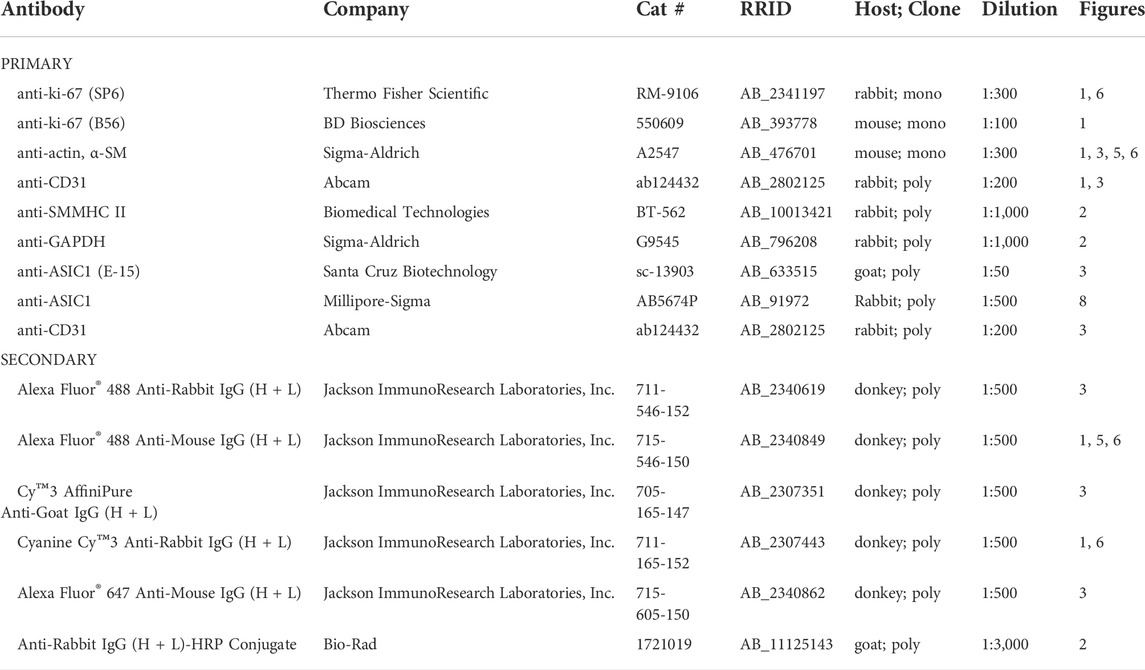 TABELLE 2. Liste der für die Immunfluoreszenz- und Western-Blot-Analyse verwendeten primären und sekundären Antikörper. Bewertung der zellulären Proliferation mittels Ki-67Lungenschnitte wurden mit Anti-Ki-67 inkubiert (
Tabelle 2
) und der Prozentsatz Ki-67-positiver SM und ECs wurde aus ca. 15–20 Gefäßen pro Tier (5 Tiere/Gruppe) mithilfe der Software ImageJ (National Institutes of Health) berechnet. Gefäße wurden anhand der Morphologie und SMA- oder CD31-Immunfluoreszenz identifiziert und Kerne wurden vor dem Aufbringen der Schnitte 15 Minuten lang bei Raumtemperatur mit TO-PRO™-3-Jodid (1:1.000; Invitrogen, T3605) gefärbt.Bewertung der zellspezifischen ASIC1-DeletionLungenschnitte wurden mit Antikörpern gegen ASIC1, SMA und CD31 inkubiert (
Tabelle 2
). Es wurden Bilder von fünf Lungenarterien pro Gruppe aufgenommen. Mithilfe der Software ImageJ (NIH) wurde eine Maske der SMA- oder CD31-Immunfluoreszenz erstellt und die mittlere Intensität von ASIC1 in jeder Maske bestimmt.Beurteilung der arteriellen Umgestaltung mittels α-SM-Aktin-ImmunfluoreszenzDie Schwellenwerte für die Bilder wurden mithilfe der Software ImageJ ermittelt. Um jede vollständig muskularisierte Arterie wurden Interessensbereiche (ROIs) gezeichnet. Der Prozentsatz des Schwellenwertbereichs im Vergleich zum gesamten ROI-Bereich wurde für jede Arterie berechnet und mit 100 multipliziert, um den Prozentsatz der Muskularisierung zu erhalten. Der Arteriendurchmesser wurde anhand des Umfangs des ROI berechnet und die Analyse wurde nach Arteriendurchmesser durchgeführt: <25 μm, 25–50 μm oder 50–100 μm. Fluoreszenzbilder wurden digital invertiert, um einen besseren Kontrast und eine bessere Sichtbarkeit der Immunfluoreszenz zu erzielen.Western-Blot-AnalyseDie Expression der SM-Myosin-Schwerkette (MHC) und des GAPDH-Proteins wurde durch Western-Blot-Analyse ermittelt. Die gesamte Lunge wurde in Tris-HCl-Homogenisierungspuffer (enthaltend 225 mM Saccharose, 2 mM Tris-HCL, 2 mM EDTA, 12 µM Leupeptin, 1 µM Pepstatin A und 0,3 µM Aprotinin) mit einem Glashomogenisator homogenisiert und 10 Minuten bei 4 °C bei 10.000 g zentrifugiert, um unlösliche Rückstände zu entfernen. Die Proteinkonzentrationen der Proben wurden durch den Qubit-Protein-Assay (Life Technologies) bestimmt. Die Proben wurden 5 Minuten in Probenpuffer gekocht und 20 µg Protein wurden durch SDS-PAGE (7,5 % Tris/Glycin) getrennt und auf eine Polyvinylidenfluoridmembran übertragen. Der Blot wurde bei Raumtemperatur 1 Stunde lang mit 5 % Magermilchpulver blockiert, dann über Nacht bei 4 °C in Primärantikörpern inkubiert und anschließend 1 Stunde lang bei Raumtemperatur in Sekundärantikörpern (
Tabelle 2
). Die Proteine wurden dann nach Chemilumineszenzmarkierung (ECL; Pierce, 32209) durch Autoradiographiefilm (GeneMate) nachgewiesen. Die Quantifizierung der Proteinexpression erfolgte mit der Software ImageJ und die MHC-Expression wurde auf GAPDH normalisiert. GAPDH wurde anschließend auf MHC getestet.Beurteilung der pulmonalen HypertonieNach 6 Wochen CH wurden die Mäuse anästhesiert (Gasgemisch aus 2 % Isofluran und 98 % O2 ) und der rechtsventrikuläre systolische Druck (RVSP) und die Herzfrequenz wurden mittels transdiaphragmatischer direkter Herzpunktion gemessen, wie zuvor beschrieben (
Nitta et al., 2014
). Um das Zwerchfell freizulegen, wurde eine obere transversale Laparotomie durchgeführt. Eine 25-Gauge-Nadel, die über einen mit Kochsalzlösung gefüllten Katheter mit einem Druckwandler (Modell APT300, Harvard Apparatus) verbunden war, wurde über einen transdiaphragmatischen Zugang bei geschlossenem Brustkorb in den RV eingeführt und die Ausgabe mit einem TAM-A-Brückenverstärker (Hugo Saks Electronik, Harvard Apparatus) verstärkt und mithilfe der Powerlab-Datenerfassung und der LabChart-Software (ADInstruments) aufgezeichnet. Die Ableitung des maximalen RV-Drucks nach der Zeit (dP/dtmax ) liefert einen Index der RV-Kontraktilität. Der Druck-Zeit-Index ist die Fläche unter der systolischen Druckkurve und gibt Aufschluss über die RV-Arbeitsbelastung und den Sauerstoffverbrauch. Die rechtsventrikuläre Hypertrophie als Reaktion auf CH wurde durch Messung des Massenverhältnisses des rechten Ventrikels zum linken Ventrikel plus Septum (Fulton-Index) beurteilt.Proliferation/Migration in PASMCsErzeugung von Maus-PASMCs (mPASMC)Die Tiere wurden mit Natriumpentobarbital (200 mg/kg Körpergewicht, IP) anästhesiert und Herz und Lunge durch eine Mittellinienthorakotomie entfernt. Intrapulmonale Arterien (∼zweite bis fünfte Ordnung) wurden vom umgebenden Lungenparenchym abgetrennt und enzymatisch verdaut, indem sie 20 Minuten lang bei 37 °C in Ca 2+ -reduzierter Hank's Balanced Salt Solution (HBSS) mit Papain (9,5 U/ml), Typ-I-Kollagenase (2 mg/ml), Dithiothreitol (1 mg/ml) und BSA (2 mg/ml) inkubiert wurden. PASMCs wurden durch vorsichtiges Verreiben mit einer feuerpolierten Pipette in Ca 2+ -freier HBSS dispergiert. Frisch dispergierte PASMCs wurden auf gelatinebeschichteten Schalen plattiert und in SM-Zellmedium (Cell Biologics) mit 10 % fötalem Rinderserum und 1 % Penicillin/Streptomycin in einer befeuchteten Atmosphäre aus 5 % CO2 -95 % Luft bei 37 °C kultiviert . Vor den Experimenten wurden PASMCs mindestens 48 Stunden lang in einem serumfreien SMC-Medium mit Insulin, EGF, Hydrocortison, L -Glutamin und 1 % Penicillin/Streptomycin (M2268SF Cell Biologics) kultiviert. Die Zellreinheit betrug >95 %, wie anhand des morphologischen Erscheinungsbilds unter Phasenkontrastmikroskopie und Immunfluoreszenzfärbung für SM 22α wie zuvor beschrieben (
Detweiler et al., 2019
) beurteilt wurde.mPASMC-ProliferationUm die Beteiligung von ASIC1a an der Proliferation zu bestimmen, wurden mPASMCs von Asic1a +/+ und Asic1a −/− Mäusen 24, 48 und 72 Stunden lang unter Hypoxie (2 % O 2 , 5 % CO 2 ) mit Bromdesoxyuridin (BrdU; 10 μM) inkubiert, wobei eine hypoxische Inkubator-Unterkammer (Biospherix C-Chamber) verwendet wurde. mPASMCs wurden fixiert und mit einem konjugierten Anti-BrdU-FITC-Antikörper (BD Biosciences) markiert, um den Einbau von BrdU in neu synthetisierte DNA von 20.000 Ereignissen pro Probe mittels Durchflusszytometrie (LSR-Fortessa-Durchflusszytometer mit FACSDiva, Softwareversion 3.0; BD Biosciences) zu messen. PDGF-BB (20 ng/ml, Millipore) wurde 72 Stunden lang hinzugefügt, um eine positive Färbekontrolle zu erzeugen.mPASMC-MigrationDie Migration von mPASMC wurde mithilfe einer modifizierten Boyden-Kammer (Costar Transwell-Einsätze, 6,5 mm Durchmesser, 8,0 µm Porengröße) beurteilt. mPASMCs wurden mithilfe eines Standard-Gittertests gezählt und mit 1 × 10 5 Zellen/Vertiefung in Basalmedium (plus 1 % FBS) auf dem Einsatz plattiert. Basalmedium wurde auch in die untere Vertiefung der Boyden-Kammer gegeben und die Zellen wurden 24 h in Normoxie (95 % Luft, 5 % CO 2 ) oder Hypoxie (2 % O 2 , 5 % CO 2 ) inkubiert, um die Migration zu stimulieren. Nach 24 h wurden mPASMCs 15 min lang mit 2 % Paraformaldehyd fixiert und dann 5 min lang mit Coomassie-Blau gefärbt. Die Zellen wurden mehrmals gewaschen, um überschüssiges Coomassie zu entfernen. Die Bilder wurden mit einem 20-fachen Objektiv auf einem Eclipse E400-Mikroskop mit einer DS-Fi1-Kamera aufgenommen und mit der Software NIS-Elements F 3.0 (Nikon) analysiert. Pro Vertiefung wurden fünf zufällige Hellfeldbilder für die Gesamtzahl der Zellen aufgenommen, bevor die nicht migrierten PASMCs von der Oberseite der Filter mit einem Wattestäbchen entfernt und weitere fünf Bilder aufgenommen wurden, um die Anzahl der migrierten Zellen zu ermitteln. Die mit Coomassie gefärbten mPASMCs wurden verwendet, um den Bereich der migrierten im Verhältnis zur Gesamtzahl der mPASMCs zu bestimmen. Dieser wurde mit 100 multipliziert, um den Prozentsatz der migrierten mPASMCs zu erhalten (ImageJ).Menschliche PASMCs (hPASMC; Cascade Biologics, #C-009-5C) wurden auf mit Poly- L -Lysin beschichteten Platten in Medium 231 (Invitrogen, #M231500) mit SM-Wachstumszusatz (Invitrogen, #S00725) in einer befeuchteten Atmosphäre aus 5 % CO2 -95 % Luft bei 37 °C gezüchtet. hPASMC wurden in Passage drei bis fünf verwendet und für die Dauer der folgenden Experimente mit Vehikel (H2O ) , Amilorid (30 μM, Enzo Life Sciences) oder Psalmotoxin 1 (PcTX1; 20 nM, Phoenix Pharmaceuticals) behandelt.ASIC1-Expression in hPASMCDie Gesamt- und Zelloberflächenexpression von ASIC1 wurde durch Western-Blot-Analyse bestimmt (siehe oben). hPASMC wurden in 75-cm2-Kolben bis zu einer Konfluenz von ungefähr 90 % gezüchtet und 12 h Normoxie (95 % Luft, 5 % CO2 ) oder Hypoxie (2 % O2 , 5 % CO2 ) ausgesetzt . Zur Bestimmung der Plasmamembranlokalisierung von ASIC1 verwendeten wir ein Zelloberflächenprotein-Isolationskit (Pierce, Thermo Fisher Scientific), wie zuvor beschrieben (
Herbert et al., 2016
,
2018
). hPASMCs wurden 30 min bei 4 °C mit Sulfo-NHS-SS-Biotin (Pierce) inkubiert. Die Reaktion wurde abgeschreckt und die hPASMCs wurden geerntet und mit 10 mM TrisHCl-Homogenisierungspuffer lysiert und 2 min bei 10.000 g zentrifugiert. Das geklärte Überstand wurde für 1 h bei Raumtemperatur zu NeutrAvidin-Agarose-Harzsäulen hinzugefügt. Der Durchfluss wurde als zytosolische Proteinfraktion gesammelt und Oberflächenprotein wurde durch Elution mit 5× Probenpuffer gesammelt. Wir haben zuvor die Spezifität des Zelloberflächenassays zur Fraktionierung von Zelloberflächen- gegenüber intrazellulären Proteinen nachgewiesen (
Herbert et al., 2016
). Oberflächenprotein (25 μl) oder zytosolische Proteinlysate (20 μg) wurden durch SDS-PAGE (7,5 % Tris/Glycin) getrennt und auf PVDF-Membranen übertragen. ASIC1 wurde in Zelloberflächen- und zytosolischen Fraktionen durch Exposition des Blots gegenüber chemilumineszenzempfindlichem Film (GeneMate) nachgewiesen. Die Quantifizierung der ASIC1-Bänder erfolgte durch densitometrische Analyse gescannter Bilder (ImageJ) und wurde als Verhältnis von Plasmamembran- zu zytosolischen densitometrischen Einheiten ausgedrückt.hPASMC-MigrationEin In-vitro- Kratztest wurde an konfluenten Monoschichten von hPASMCs durchgeführt. Die Monoschichten wurden manuell mit der Spitze einer 100-µl-Pipette abgekratzt und dann vorsichtig zweimal mit PBS gespült, um nicht anhaftende Zellen zu entfernen. Bilder der verletzten Stelle wurden unmittelbar nach dem Kratzer (Zeitpunkt Null) und nach einer 12-stündigen Exposition gegenüber Normoxie (95 % Luft, 5 % CO2 ) oder Hypoxie (2 % O2 , 5 % CO2 ) aufgenommen. Die Bilder wurden mit einem 20-fachen Objektiv auf einem Eclipse E400-Mikroskop mit einer DS-Fi1-Kamera aufgenommen und mit der Software NIS-Elements F 3.0 (Nikon) analysiert. Ein an der Unterseite der Zellkulturplatte angebrachtes Gitter diente als Referenzpunkt, um in jedem Zeitintervall Bilder derselben Stelle aufzunehmen. Die verletzte Stelle wurde mit ImageJ (National Institutes of Health) bestimmt. Die Heilung wurde quantifiziert als % Reinvasion = (AreaI–AreaT)/AreaI × 100 %, wobei: AreaI = Anfangsbereich und AreaT = Bereich zum Zeitpunkt (T) 12 Stunden nach der Verletzung.hPASMC-VermehrunghPASMCs wurden trypsiniert und die Zellsuspension wurde mit gleichen Teilen Trypanblaulösung bis zu einer Endkonzentration von 0,4 % gemischt, um die Zelllebensfähigkeit zu beurteilen. Eine homogene Mischung wurde in eine Einwegkammer geladen und die Zellzahl wurde mithilfe des automatischen Countess-Zellzählers (Invitrogen) bestimmt.StatistikenAlle Daten werden als Mittelwert ± Standardfehler ausgedrückt. Prozentuale Daten wurden vor der parametrischen Analyse durch Arkussinustransformationen in Normalverteilungen umgewandelt. Die Normalverteilung wurde mit dem Shapiro-Wilks-Normalitätstest ( p > 0,05) getestet. Die Werte von n und die statistischen Tests sind in den Bildlegenden angegeben und wurden mit Prism 9 (GraphPad Software) durchgeführt. Eine Wahrscheinlichkeit von ≤ 0,05 mit einem Leistungsniveau von 0,80 wurde für alle Vergleiche als statistisch signifikant akzeptiert.ErgebnisseCH-induzierte vaskuläre Zellproliferation und phänotypischer Wechsel sind ASIC1a-abhängigKi-67 ist ein Kernprotein, das während der Zellproliferation exprimiert wird. Um die In-vivo- Proliferation von Gefäßzellen im Verlauf der Entwicklung einer durch CH induzierten pulmonalen Hypertonie zu untersuchen, ermittelten wir den Prozentsatz Ki-67-positiver PAECs und PASMCs nach 0-, 3-, 7- und 28-tägiger CH-Exposition in Asic1a +/+- Mäusen (
Abbildung 1A
). Die Zahl proliferierender PAECs und PASMCs war nach 3 Tagen CH am höchsten (
Abbildung 1B
). Der Prozentsatz proliferierender PAECs und PASMCs war nach 7 Tagen immer noch erhöht, aber die PASMC-Proliferation nahm um die Hälfte ab. Der Prozentsatz proliferierender PAECs und PASMCs nach 28 Tagen unterschied sich nicht signifikant vom Ausgangswert (
Abbildung 1B
), da die Mehrheit der proliferierenden Zellen nach 28 Tagen extravaskuläre Zellen waren. Basierend auf diesen Daten untersuchten wir dann die PAEC- und PASMC-Proliferation in Asic1a −/-- Mäusen nach 3 Tagen CH. Der Prozentsatz proliferierender PAECs war bei Asic1a -/- Mäusen signifikant reduziert, im Vergleich zu Kontrollmäusen jedoch erhöht (
Abbildung 1C
). Darüber hinaus hatte CH keinen Effekt auf die Proliferation von PASMCs bei Asic1a -/- Mäusen (
Abbildung 1D
). Abbildung 1
TABELLE 2. Liste der für die Immunfluoreszenz- und Western-Blot-Analyse verwendeten primären und sekundären Antikörper. Bewertung der zellulären Proliferation mittels Ki-67Lungenschnitte wurden mit Anti-Ki-67 inkubiert (
Tabelle 2
) und der Prozentsatz Ki-67-positiver SM und ECs wurde aus ca. 15–20 Gefäßen pro Tier (5 Tiere/Gruppe) mithilfe der Software ImageJ (National Institutes of Health) berechnet. Gefäße wurden anhand der Morphologie und SMA- oder CD31-Immunfluoreszenz identifiziert und Kerne wurden vor dem Aufbringen der Schnitte 15 Minuten lang bei Raumtemperatur mit TO-PRO™-3-Jodid (1:1.000; Invitrogen, T3605) gefärbt.Bewertung der zellspezifischen ASIC1-DeletionLungenschnitte wurden mit Antikörpern gegen ASIC1, SMA und CD31 inkubiert (
Tabelle 2
). Es wurden Bilder von fünf Lungenarterien pro Gruppe aufgenommen. Mithilfe der Software ImageJ (NIH) wurde eine Maske der SMA- oder CD31-Immunfluoreszenz erstellt und die mittlere Intensität von ASIC1 in jeder Maske bestimmt.Beurteilung der arteriellen Umgestaltung mittels α-SM-Aktin-ImmunfluoreszenzDie Schwellenwerte für die Bilder wurden mithilfe der Software ImageJ ermittelt. Um jede vollständig muskularisierte Arterie wurden Interessensbereiche (ROIs) gezeichnet. Der Prozentsatz des Schwellenwertbereichs im Vergleich zum gesamten ROI-Bereich wurde für jede Arterie berechnet und mit 100 multipliziert, um den Prozentsatz der Muskularisierung zu erhalten. Der Arteriendurchmesser wurde anhand des Umfangs des ROI berechnet und die Analyse wurde nach Arteriendurchmesser durchgeführt: <25 μm, 25–50 μm oder 50–100 μm. Fluoreszenzbilder wurden digital invertiert, um einen besseren Kontrast und eine bessere Sichtbarkeit der Immunfluoreszenz zu erzielen.Western-Blot-AnalyseDie Expression der SM-Myosin-Schwerkette (MHC) und des GAPDH-Proteins wurde durch Western-Blot-Analyse ermittelt. Die gesamte Lunge wurde in Tris-HCl-Homogenisierungspuffer (enthaltend 225 mM Saccharose, 2 mM Tris-HCL, 2 mM EDTA, 12 µM Leupeptin, 1 µM Pepstatin A und 0,3 µM Aprotinin) mit einem Glashomogenisator homogenisiert und 10 Minuten bei 4 °C bei 10.000 g zentrifugiert, um unlösliche Rückstände zu entfernen. Die Proteinkonzentrationen der Proben wurden durch den Qubit-Protein-Assay (Life Technologies) bestimmt. Die Proben wurden 5 Minuten in Probenpuffer gekocht und 20 µg Protein wurden durch SDS-PAGE (7,5 % Tris/Glycin) getrennt und auf eine Polyvinylidenfluoridmembran übertragen. Der Blot wurde bei Raumtemperatur 1 Stunde lang mit 5 % Magermilchpulver blockiert, dann über Nacht bei 4 °C in Primärantikörpern inkubiert und anschließend 1 Stunde lang bei Raumtemperatur in Sekundärantikörpern (
Tabelle 2
). Die Proteine wurden dann nach Chemilumineszenzmarkierung (ECL; Pierce, 32209) durch Autoradiographiefilm (GeneMate) nachgewiesen. Die Quantifizierung der Proteinexpression erfolgte mit der Software ImageJ und die MHC-Expression wurde auf GAPDH normalisiert. GAPDH wurde anschließend auf MHC getestet.Beurteilung der pulmonalen HypertonieNach 6 Wochen CH wurden die Mäuse anästhesiert (Gasgemisch aus 2 % Isofluran und 98 % O2 ) und der rechtsventrikuläre systolische Druck (RVSP) und die Herzfrequenz wurden mittels transdiaphragmatischer direkter Herzpunktion gemessen, wie zuvor beschrieben (
Nitta et al., 2014
). Um das Zwerchfell freizulegen, wurde eine obere transversale Laparotomie durchgeführt. Eine 25-Gauge-Nadel, die über einen mit Kochsalzlösung gefüllten Katheter mit einem Druckwandler (Modell APT300, Harvard Apparatus) verbunden war, wurde über einen transdiaphragmatischen Zugang bei geschlossenem Brustkorb in den RV eingeführt und die Ausgabe mit einem TAM-A-Brückenverstärker (Hugo Saks Electronik, Harvard Apparatus) verstärkt und mithilfe der Powerlab-Datenerfassung und der LabChart-Software (ADInstruments) aufgezeichnet. Die Ableitung des maximalen RV-Drucks nach der Zeit (dP/dtmax ) liefert einen Index der RV-Kontraktilität. Der Druck-Zeit-Index ist die Fläche unter der systolischen Druckkurve und gibt Aufschluss über die RV-Arbeitsbelastung und den Sauerstoffverbrauch. Die rechtsventrikuläre Hypertrophie als Reaktion auf CH wurde durch Messung des Massenverhältnisses des rechten Ventrikels zum linken Ventrikel plus Septum (Fulton-Index) beurteilt.Proliferation/Migration in PASMCsErzeugung von Maus-PASMCs (mPASMC)Die Tiere wurden mit Natriumpentobarbital (200 mg/kg Körpergewicht, IP) anästhesiert und Herz und Lunge durch eine Mittellinienthorakotomie entfernt. Intrapulmonale Arterien (∼zweite bis fünfte Ordnung) wurden vom umgebenden Lungenparenchym abgetrennt und enzymatisch verdaut, indem sie 20 Minuten lang bei 37 °C in Ca 2+ -reduzierter Hank's Balanced Salt Solution (HBSS) mit Papain (9,5 U/ml), Typ-I-Kollagenase (2 mg/ml), Dithiothreitol (1 mg/ml) und BSA (2 mg/ml) inkubiert wurden. PASMCs wurden durch vorsichtiges Verreiben mit einer feuerpolierten Pipette in Ca 2+ -freier HBSS dispergiert. Frisch dispergierte PASMCs wurden auf gelatinebeschichteten Schalen plattiert und in SM-Zellmedium (Cell Biologics) mit 10 % fötalem Rinderserum und 1 % Penicillin/Streptomycin in einer befeuchteten Atmosphäre aus 5 % CO2 -95 % Luft bei 37 °C kultiviert . Vor den Experimenten wurden PASMCs mindestens 48 Stunden lang in einem serumfreien SMC-Medium mit Insulin, EGF, Hydrocortison, L -Glutamin und 1 % Penicillin/Streptomycin (M2268SF Cell Biologics) kultiviert. Die Zellreinheit betrug >95 %, wie anhand des morphologischen Erscheinungsbilds unter Phasenkontrastmikroskopie und Immunfluoreszenzfärbung für SM 22α wie zuvor beschrieben (
Detweiler et al., 2019
) beurteilt wurde.mPASMC-ProliferationUm die Beteiligung von ASIC1a an der Proliferation zu bestimmen, wurden mPASMCs von Asic1a +/+ und Asic1a −/− Mäusen 24, 48 und 72 Stunden lang unter Hypoxie (2 % O 2 , 5 % CO 2 ) mit Bromdesoxyuridin (BrdU; 10 μM) inkubiert, wobei eine hypoxische Inkubator-Unterkammer (Biospherix C-Chamber) verwendet wurde. mPASMCs wurden fixiert und mit einem konjugierten Anti-BrdU-FITC-Antikörper (BD Biosciences) markiert, um den Einbau von BrdU in neu synthetisierte DNA von 20.000 Ereignissen pro Probe mittels Durchflusszytometrie (LSR-Fortessa-Durchflusszytometer mit FACSDiva, Softwareversion 3.0; BD Biosciences) zu messen. PDGF-BB (20 ng/ml, Millipore) wurde 72 Stunden lang hinzugefügt, um eine positive Färbekontrolle zu erzeugen.mPASMC-MigrationDie Migration von mPASMC wurde mithilfe einer modifizierten Boyden-Kammer (Costar Transwell-Einsätze, 6,5 mm Durchmesser, 8,0 µm Porengröße) beurteilt. mPASMCs wurden mithilfe eines Standard-Gittertests gezählt und mit 1 × 10 5 Zellen/Vertiefung in Basalmedium (plus 1 % FBS) auf dem Einsatz plattiert. Basalmedium wurde auch in die untere Vertiefung der Boyden-Kammer gegeben und die Zellen wurden 24 h in Normoxie (95 % Luft, 5 % CO 2 ) oder Hypoxie (2 % O 2 , 5 % CO 2 ) inkubiert, um die Migration zu stimulieren. Nach 24 h wurden mPASMCs 15 min lang mit 2 % Paraformaldehyd fixiert und dann 5 min lang mit Coomassie-Blau gefärbt. Die Zellen wurden mehrmals gewaschen, um überschüssiges Coomassie zu entfernen. Die Bilder wurden mit einem 20-fachen Objektiv auf einem Eclipse E400-Mikroskop mit einer DS-Fi1-Kamera aufgenommen und mit der Software NIS-Elements F 3.0 (Nikon) analysiert. Pro Vertiefung wurden fünf zufällige Hellfeldbilder für die Gesamtzahl der Zellen aufgenommen, bevor die nicht migrierten PASMCs von der Oberseite der Filter mit einem Wattestäbchen entfernt und weitere fünf Bilder aufgenommen wurden, um die Anzahl der migrierten Zellen zu ermitteln. Die mit Coomassie gefärbten mPASMCs wurden verwendet, um den Bereich der migrierten im Verhältnis zur Gesamtzahl der mPASMCs zu bestimmen. Dieser wurde mit 100 multipliziert, um den Prozentsatz der migrierten mPASMCs zu erhalten (ImageJ).Menschliche PASMCs (hPASMC; Cascade Biologics, #C-009-5C) wurden auf mit Poly- L -Lysin beschichteten Platten in Medium 231 (Invitrogen, #M231500) mit SM-Wachstumszusatz (Invitrogen, #S00725) in einer befeuchteten Atmosphäre aus 5 % CO2 -95 % Luft bei 37 °C gezüchtet. hPASMC wurden in Passage drei bis fünf verwendet und für die Dauer der folgenden Experimente mit Vehikel (H2O ) , Amilorid (30 μM, Enzo Life Sciences) oder Psalmotoxin 1 (PcTX1; 20 nM, Phoenix Pharmaceuticals) behandelt.ASIC1-Expression in hPASMCDie Gesamt- und Zelloberflächenexpression von ASIC1 wurde durch Western-Blot-Analyse bestimmt (siehe oben). hPASMC wurden in 75-cm2-Kolben bis zu einer Konfluenz von ungefähr 90 % gezüchtet und 12 h Normoxie (95 % Luft, 5 % CO2 ) oder Hypoxie (2 % O2 , 5 % CO2 ) ausgesetzt . Zur Bestimmung der Plasmamembranlokalisierung von ASIC1 verwendeten wir ein Zelloberflächenprotein-Isolationskit (Pierce, Thermo Fisher Scientific), wie zuvor beschrieben (
Herbert et al., 2016
,
2018
). hPASMCs wurden 30 min bei 4 °C mit Sulfo-NHS-SS-Biotin (Pierce) inkubiert. Die Reaktion wurde abgeschreckt und die hPASMCs wurden geerntet und mit 10 mM TrisHCl-Homogenisierungspuffer lysiert und 2 min bei 10.000 g zentrifugiert. Das geklärte Überstand wurde für 1 h bei Raumtemperatur zu NeutrAvidin-Agarose-Harzsäulen hinzugefügt. Der Durchfluss wurde als zytosolische Proteinfraktion gesammelt und Oberflächenprotein wurde durch Elution mit 5× Probenpuffer gesammelt. Wir haben zuvor die Spezifität des Zelloberflächenassays zur Fraktionierung von Zelloberflächen- gegenüber intrazellulären Proteinen nachgewiesen (
Herbert et al., 2016
). Oberflächenprotein (25 μl) oder zytosolische Proteinlysate (20 μg) wurden durch SDS-PAGE (7,5 % Tris/Glycin) getrennt und auf PVDF-Membranen übertragen. ASIC1 wurde in Zelloberflächen- und zytosolischen Fraktionen durch Exposition des Blots gegenüber chemilumineszenzempfindlichem Film (GeneMate) nachgewiesen. Die Quantifizierung der ASIC1-Bänder erfolgte durch densitometrische Analyse gescannter Bilder (ImageJ) und wurde als Verhältnis von Plasmamembran- zu zytosolischen densitometrischen Einheiten ausgedrückt.hPASMC-MigrationEin In-vitro- Kratztest wurde an konfluenten Monoschichten von hPASMCs durchgeführt. Die Monoschichten wurden manuell mit der Spitze einer 100-µl-Pipette abgekratzt und dann vorsichtig zweimal mit PBS gespült, um nicht anhaftende Zellen zu entfernen. Bilder der verletzten Stelle wurden unmittelbar nach dem Kratzer (Zeitpunkt Null) und nach einer 12-stündigen Exposition gegenüber Normoxie (95 % Luft, 5 % CO2 ) oder Hypoxie (2 % O2 , 5 % CO2 ) aufgenommen. Die Bilder wurden mit einem 20-fachen Objektiv auf einem Eclipse E400-Mikroskop mit einer DS-Fi1-Kamera aufgenommen und mit der Software NIS-Elements F 3.0 (Nikon) analysiert. Ein an der Unterseite der Zellkulturplatte angebrachtes Gitter diente als Referenzpunkt, um in jedem Zeitintervall Bilder derselben Stelle aufzunehmen. Die verletzte Stelle wurde mit ImageJ (National Institutes of Health) bestimmt. Die Heilung wurde quantifiziert als % Reinvasion = (AreaI–AreaT)/AreaI × 100 %, wobei: AreaI = Anfangsbereich und AreaT = Bereich zum Zeitpunkt (T) 12 Stunden nach der Verletzung.hPASMC-VermehrunghPASMCs wurden trypsiniert und die Zellsuspension wurde mit gleichen Teilen Trypanblaulösung bis zu einer Endkonzentration von 0,4 % gemischt, um die Zelllebensfähigkeit zu beurteilen. Eine homogene Mischung wurde in eine Einwegkammer geladen und die Zellzahl wurde mithilfe des automatischen Countess-Zellzählers (Invitrogen) bestimmt.StatistikenAlle Daten werden als Mittelwert ± Standardfehler ausgedrückt. Prozentuale Daten wurden vor der parametrischen Analyse durch Arkussinustransformationen in Normalverteilungen umgewandelt. Die Normalverteilung wurde mit dem Shapiro-Wilks-Normalitätstest ( p > 0,05) getestet. Die Werte von n und die statistischen Tests sind in den Bildlegenden angegeben und wurden mit Prism 9 (GraphPad Software) durchgeführt. Eine Wahrscheinlichkeit von ≤ 0,05 mit einem Leistungsniveau von 0,80 wurde für alle Vergleiche als statistisch signifikant akzeptiert.ErgebnisseCH-induzierte vaskuläre Zellproliferation und phänotypischer Wechsel sind ASIC1a-abhängigKi-67 ist ein Kernprotein, das während der Zellproliferation exprimiert wird. Um die In-vivo- Proliferation von Gefäßzellen im Verlauf der Entwicklung einer durch CH induzierten pulmonalen Hypertonie zu untersuchen, ermittelten wir den Prozentsatz Ki-67-positiver PAECs und PASMCs nach 0-, 3-, 7- und 28-tägiger CH-Exposition in Asic1a +/+- Mäusen (
Abbildung 1A
). Die Zahl proliferierender PAECs und PASMCs war nach 3 Tagen CH am höchsten (
Abbildung 1B
). Der Prozentsatz proliferierender PAECs und PASMCs war nach 7 Tagen immer noch erhöht, aber die PASMC-Proliferation nahm um die Hälfte ab. Der Prozentsatz proliferierender PAECs und PASMCs nach 28 Tagen unterschied sich nicht signifikant vom Ausgangswert (
Abbildung 1B
), da die Mehrheit der proliferierenden Zellen nach 28 Tagen extravaskuläre Zellen waren. Basierend auf diesen Daten untersuchten wir dann die PAEC- und PASMC-Proliferation in Asic1a −/-- Mäusen nach 3 Tagen CH. Der Prozentsatz proliferierender PAECs war bei Asic1a -/- Mäusen signifikant reduziert, im Vergleich zu Kontrollmäusen jedoch erhöht (
Abbildung 1C
). Darüber hinaus hatte CH keinen Effekt auf die Proliferation von PASMCs bei Asic1a -/- Mäusen (
Abbildung 1D
). Abbildung 1
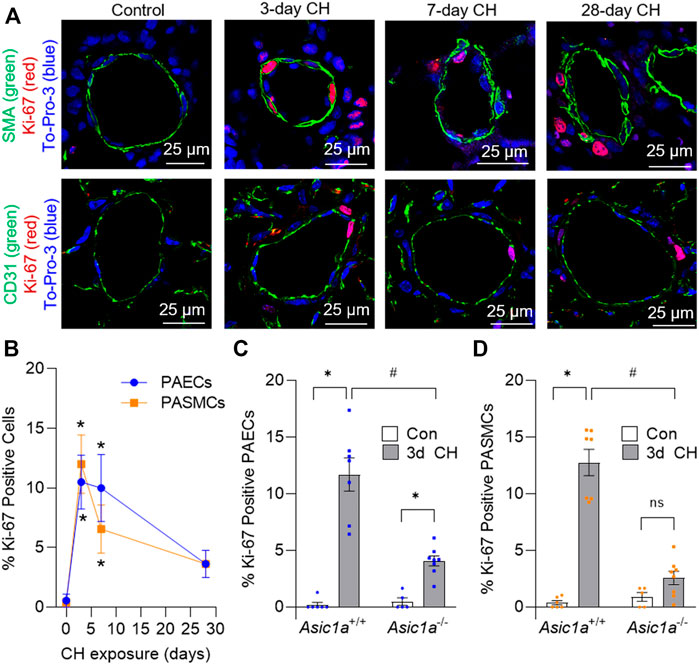 ABBILDUNG 1. Die durch CH induzierte Proliferation von Gefäßzellen ist ASIC1a-abhängig. (A) Repräsentative Immunfluoreszenzbilder, die Ki-67 (rot), α-SMA (grün, obere Reihe), CD31 (grün, untere Reihe) und To-Pro-3 (blau) in kleinen Lungenarterien (<100 µm) von Asic1a +/+ Mäusen zeigen, und (
ABBILDUNG 1. Die durch CH induzierte Proliferation von Gefäßzellen ist ASIC1a-abhängig. (A) Repräsentative Immunfluoreszenzbilder, die Ki-67 (rot), α-SMA (grün, obere Reihe), CD31 (grün, untere Reihe) und To-Pro-3 (blau) in kleinen Lungenarterien (<100 µm) von Asic1a +/+ Mäusen zeigen, und (") zusammenfassende Daten, die den Prozentsatz Ki-67-positiver pulmonaler arterieller Endothelzellen (PAECs, blaue Kreise) und pulmonaler arterieller glatter Muskelzellen (PASMCs, orange Quadrate) unter Kontrollbedingungen oder nach Exposition gegenüber CH (3, 7 oder 28 Tage) zeigen. n = 3 Tiere pro Gruppe (für jedes Tier wurden im Durchschnitt ca. 20 Gefäße ermittelt); analysiert als einfaktorielle ANOVA für jeden Zelltyp und einzelne Gruppen verglichen mit Šídáks Mehrfachvergleichstests. Zusammenfassung der Daten, die den Prozentsatz Ki-67-positiver (C) PAECs und (D) PASMCs in kleinen Lungenarterien (<100 µm) von Asic1a +/+ und Asic1a −/- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition zeigen. n = fünf bis acht Tiere (durchschnittlich ∼15 Gefäße pro Tier); Analyse mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für PAECs und PASMCs) wurden mit Šídáks Mehrfachvergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. entsprechende 3-tägige CH; ns = nicht signifikant. Als nächstes ermittelten wir, ob die durch CH induzierte Zunahme der PASMC-Proliferation mit einem Verlust des kontraktilen Phänotyps verbunden ist, indem wir die Proteinexpression der SM-Myosin-Schwerkette (MHC) im Lungengewebe nach 0 (Con), 1, 2, 3, 5 und 7 Tagen CH analysierten (
Abbildung 2A
). In Übereinstimmung mit dem höchsten Prozentsatz Ki-67-positiver PASMCs nach 3 Tagen CH (
Abbildung 1B
) war MHC nach 3 Tagen CH bei Asic1a +/+- Tieren signifikant erniedrigt (
Abbildung 2B, C
). Nach 7 Tagen CH war MHC im Vergleich zu den Kontrollwerten erhöht (
Abbildung 2B
). CH verringerte die MHC-Spiegel bei Asic1a −/-- Mäusen nicht (
Abbildung 2C
). Hypoxie veränderte die Expressionsniveaus von GAPDH nicht ( p = 0,2082). Zusammengenommen deuten diese Daten darauf hin, dass ASIC1a zur Proliferation von PAEC und PASMC sowie zu phänotypischen Veränderungen von PASMC beiträgt, die in den Lungenarterien als Reaktion auf CH-Exposition auftreten. Figur 2
zusammenfassende Daten, die den Prozentsatz Ki-67-positiver pulmonaler arterieller Endothelzellen (PAECs, blaue Kreise) und pulmonaler arterieller glatter Muskelzellen (PASMCs, orange Quadrate) unter Kontrollbedingungen oder nach Exposition gegenüber CH (3, 7 oder 28 Tage) zeigen. n = 3 Tiere pro Gruppe (für jedes Tier wurden im Durchschnitt ca. 20 Gefäße ermittelt); analysiert als einfaktorielle ANOVA für jeden Zelltyp und einzelne Gruppen verglichen mit Šídáks Mehrfachvergleichstests. Zusammenfassung der Daten, die den Prozentsatz Ki-67-positiver (C) PAECs und (D) PASMCs in kleinen Lungenarterien (<100 µm) von Asic1a +/+ und Asic1a −/- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition zeigen. n = fünf bis acht Tiere (durchschnittlich ∼15 Gefäße pro Tier); Analyse mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für PAECs und PASMCs) wurden mit Šídáks Mehrfachvergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. entsprechende 3-tägige CH; ns = nicht signifikant. Als nächstes ermittelten wir, ob die durch CH induzierte Zunahme der PASMC-Proliferation mit einem Verlust des kontraktilen Phänotyps verbunden ist, indem wir die Proteinexpression der SM-Myosin-Schwerkette (MHC) im Lungengewebe nach 0 (Con), 1, 2, 3, 5 und 7 Tagen CH analysierten (
Abbildung 2A
). In Übereinstimmung mit dem höchsten Prozentsatz Ki-67-positiver PASMCs nach 3 Tagen CH (
Abbildung 1B
) war MHC nach 3 Tagen CH bei Asic1a +/+- Tieren signifikant erniedrigt (
Abbildung 2B, C
). Nach 7 Tagen CH war MHC im Vergleich zu den Kontrollwerten erhöht (
Abbildung 2B
). CH verringerte die MHC-Spiegel bei Asic1a −/-- Mäusen nicht (
Abbildung 2C
). Hypoxie veränderte die Expressionsniveaus von GAPDH nicht ( p = 0,2082). Zusammengenommen deuten diese Daten darauf hin, dass ASIC1a zur Proliferation von PAEC und PASMC sowie zu phänotypischen Veränderungen von PASMC beiträgt, die in den Lungenarterien als Reaktion auf CH-Exposition auftreten. Figur 2
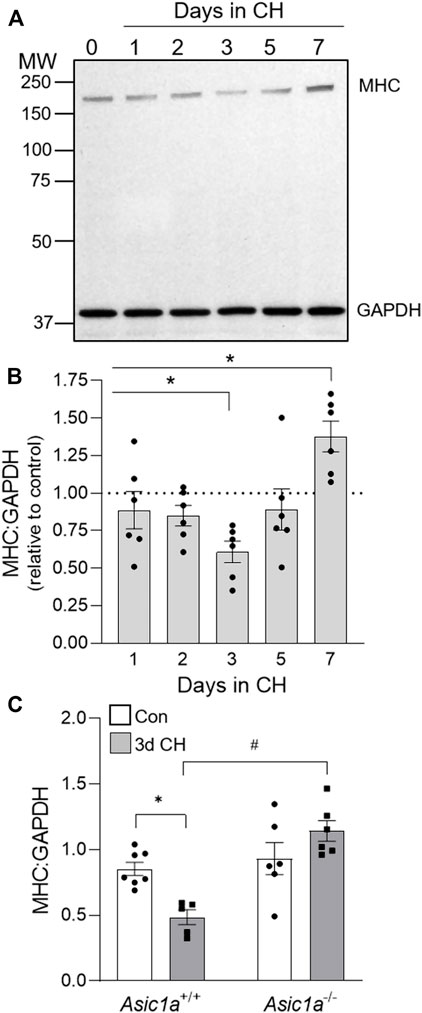 ABBILDUNG 2 . Der durch CH induzierte Verlust des kontraktilen Proteins MHC ist ASIC1a-abhängig. (A) Repräsentativer Western Blot und ( die Wirkung von CH-Exposition (1–7 Tage) auf die Expression des MHC-zu-GAPDH-Proteins in Gesamtlungenhomogenaten von Asic1a +/+- Tieren. Das gescannte Bild des Films wurde in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. n = 6/Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen wurden mit Šídáks mehrfachen Vergleichstests verglichen. (C) Zusammenfassende Daten für MHC zu GAPDH in Gesamtlungen von Asic1a +/+- und Asic1a −/-- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition. n = 5-7 Tiere/Gruppe; analysiert mit zweifaktorieller ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0022) wurden mit Šídáks mehrfachen Vergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. Asic1a +/+ Mäuse. SM-spezifisches Knockout von Asic1a schützt vor der Entwicklung von hypoxischer pulmonaler Hypertonie und kehrt diese umUm die spezifische Rolle von ASIC1a bei der Umgestaltung von PAEC und PASMC bei pulmonaler Hypertonie zu bestimmen, haben wir Mäuse mit entweder EC- (Tek Cre - Asic1a fl/fl ) oder bedingter SM-spezifischer (MHC CreER - Asic1a fl/fl ) Deletion von Asic1a erzeugt. Wie bereits zuvor bei Asic1a +/+ und Asic1a −/- Mäusen gezeigt (
Nitta et al., 2014 ), wurde ASIC1 als punktförmige Fluoreszenz in den PASMCs und PAECs von
Asic1a fl/fl und MHC CreER - Asic1a fl/fl (ohne TAM-induzierte Cre-Rekombinase) Mäusen nachgewiesen (
Abbildungen 3A,B
). Das Linienprofil durch die Gefäßwand zeigt, dass bei Tek Cre – Asic1a fl/fl- Mäusen die Expression von ASIC1 in ECs fehlt, die Expression in PASMCs jedoch erhalten bleibt, während bei MHC CreER – Asic1a fl/fl (TAM) (mit TAM-induzierter Cre-Rekombinase) die Expression von ASIC1 in PASMCs fehlt, die Expression in PAECs jedoch erhalten bleibt (
Abbildungen 3A–D
).
Abbildung 3E
zeigt die TAM-induzierte Cre-Rekombination zwischen loxP- Stellen und den Verlust der dazwischenliegenden Genomsequenz (Exons 2–3) in der Schwanz-DNA vor und nach TAM im selben Tier. ASIC1 wird im zentralen Nervensystem stark exprimiert und
Abbildung 3F zeigt, dass die TAM-induzierte Cre-Rekombinase
die Asic1a- mRNA-Werte im Hirngewebe nicht signifikant veränderte , in isolierten Pulmonalarterien (PA) jedoch keine nachweisbare Expression stattfand. Figur 3
ABBILDUNG 2 . Der durch CH induzierte Verlust des kontraktilen Proteins MHC ist ASIC1a-abhängig. (A) Repräsentativer Western Blot und ( die Wirkung von CH-Exposition (1–7 Tage) auf die Expression des MHC-zu-GAPDH-Proteins in Gesamtlungenhomogenaten von Asic1a +/+- Tieren. Das gescannte Bild des Films wurde in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. n = 6/Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen wurden mit Šídáks mehrfachen Vergleichstests verglichen. (C) Zusammenfassende Daten für MHC zu GAPDH in Gesamtlungen von Asic1a +/+- und Asic1a −/-- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition. n = 5-7 Tiere/Gruppe; analysiert mit zweifaktorieller ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0022) wurden mit Šídáks mehrfachen Vergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. Asic1a +/+ Mäuse. SM-spezifisches Knockout von Asic1a schützt vor der Entwicklung von hypoxischer pulmonaler Hypertonie und kehrt diese umUm die spezifische Rolle von ASIC1a bei der Umgestaltung von PAEC und PASMC bei pulmonaler Hypertonie zu bestimmen, haben wir Mäuse mit entweder EC- (Tek Cre - Asic1a fl/fl ) oder bedingter SM-spezifischer (MHC CreER - Asic1a fl/fl ) Deletion von Asic1a erzeugt. Wie bereits zuvor bei Asic1a +/+ und Asic1a −/- Mäusen gezeigt (
Nitta et al., 2014 ), wurde ASIC1 als punktförmige Fluoreszenz in den PASMCs und PAECs von
Asic1a fl/fl und MHC CreER - Asic1a fl/fl (ohne TAM-induzierte Cre-Rekombinase) Mäusen nachgewiesen (
Abbildungen 3A,B
). Das Linienprofil durch die Gefäßwand zeigt, dass bei Tek Cre – Asic1a fl/fl- Mäusen die Expression von ASIC1 in ECs fehlt, die Expression in PASMCs jedoch erhalten bleibt, während bei MHC CreER – Asic1a fl/fl (TAM) (mit TAM-induzierter Cre-Rekombinase) die Expression von ASIC1 in PASMCs fehlt, die Expression in PAECs jedoch erhalten bleibt (
Abbildungen 3A–D
).
Abbildung 3E
zeigt die TAM-induzierte Cre-Rekombination zwischen loxP- Stellen und den Verlust der dazwischenliegenden Genomsequenz (Exons 2–3) in der Schwanz-DNA vor und nach TAM im selben Tier. ASIC1 wird im zentralen Nervensystem stark exprimiert und
Abbildung 3F zeigt, dass die TAM-induzierte Cre-Rekombinase
die Asic1a- mRNA-Werte im Hirngewebe nicht signifikant veränderte , in isolierten Pulmonalarterien (PA) jedoch keine nachweisbare Expression stattfand. Figur 3
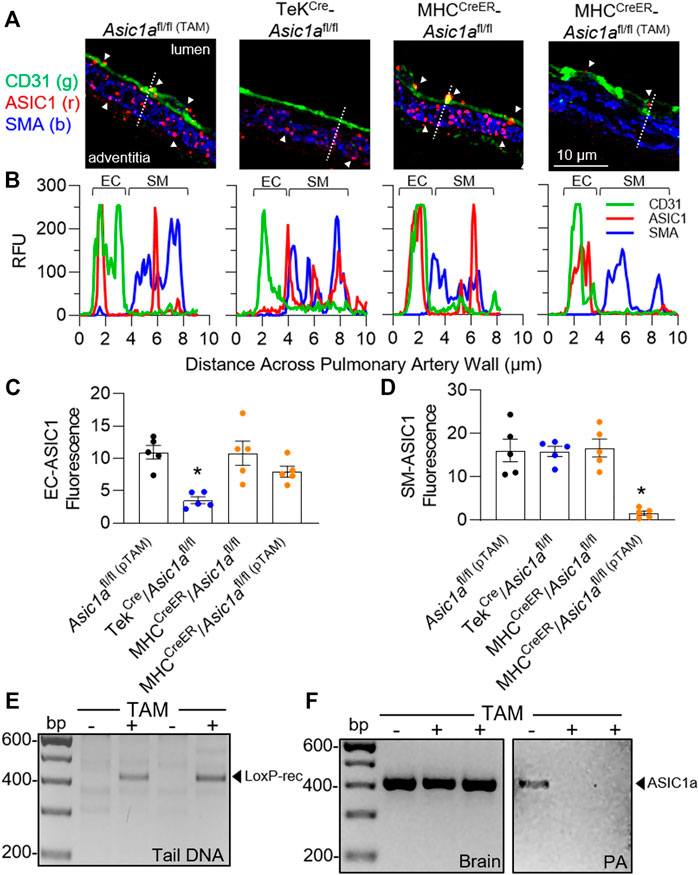 ABBILDUNG 3 . Charakterisierung transgener Mäuse. (A) Repräsentative Immunfluoreszenz für ASIC1 (rot), CD31 (grün) und SMA (blau) in Lungenarterien aus Lungenschnitten von Asic1a fl/fl (TAM) , TekCre- Asic1a fl/fl und MHC CreER - Asic1a fl/fl Mäusen, die mit Vehikel oder Tamoxifen (TAM) behandelt wurden. Weiße Pfeilspitzen zeigen punktförmige Immunfluoreszenz von ASIC1 in EC oder SM. Die gepunktete Linie in jedem Bild zeigt das in ( gezeigte Linienprofil der relativen Fluoreszenzeinheiten über die Arterienwand vom Lumen bis zur Adventitia. Zusammenfassende Analyse des ASIC1-Ausdrucks in entweder (C) EC oder (D) SM von Lungenarterien aus Lungenschnitten. n = 5 Tiere pro Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen verglichen mit Šídáks multiplen Vergleichstests. (E) PCR der Schwanz-DNA, die Cre-vermittelte LoxP-Rekombination und Exzision von gezieltem Asic1a in derselben MHC CreER - Asic1a fl/fl -Maus vor (-) und nach (+) Tamoxifen (TAM)-Behandlung zeigt. (F) Asic1a- mRNA-Expression in Hirngewebe und intrapulmonalen Arterien (PA) in MHC CreER - Asic1a fl/fl- Mäusen mit Vehikel (-) oder TAM (+). Gescannte Bilder der Blots wurden invertiert und hinsichtlich Helligkeit/Kontrast angepasst. Um die Rolle von PAEC und PASMC ASIC1a bei CH-induzierter pulmonaler Hypertonie zu bestimmen, haben wir drei verschiedene CH-Behandlungsparadigmen entwickelt, die in
Abbildung 4A
dargestellt sind : 1) Vehikel: Asic1a +/+ , Asic1a −/- , Tek Cre – Asic1a fl/fl und MHC CreER – Asic1a fl/fl Mäuse wurden mit Vehikel (Maisöl) behandelt und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 2) vorbeugend (pTAM): Asic1a fl/fl (pTAM) und MHC CreER – Asic1a fl/fl (pTAM) Mäuse wurden mit TAM behandelt (5 Tage) und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 3) therapeutisch (tTAM): MHC CreER – Asic1a fl/fl (tTAM) Mäuse wurden 3 Wochen lang einer Kontrollgruppe oder CH ausgesetzt, um pulmonale Hypertonie festzustellen. Nach 3 Wochen CH wurden die Mäuse mit TAM behandelt (5 Tage mit con/CH-Exposition) und dann weitere 2 Wochen mit con/CH behandelt.
Tabelle 3
zeigt, dass die selektive Deletion von SM- oder EC- Asic1a den mittleren arteriellen Blutdruck oder die Herzfrequenz bei wachen Mäusen nicht signifikant veränderte und dem ähnelt, was wir zuvor bei Wildtyp-Mäusen beobachtet haben (
Detweiler et al., 2019
). Figur 4
ABBILDUNG 3 . Charakterisierung transgener Mäuse. (A) Repräsentative Immunfluoreszenz für ASIC1 (rot), CD31 (grün) und SMA (blau) in Lungenarterien aus Lungenschnitten von Asic1a fl/fl (TAM) , TekCre- Asic1a fl/fl und MHC CreER - Asic1a fl/fl Mäusen, die mit Vehikel oder Tamoxifen (TAM) behandelt wurden. Weiße Pfeilspitzen zeigen punktförmige Immunfluoreszenz von ASIC1 in EC oder SM. Die gepunktete Linie in jedem Bild zeigt das in ( gezeigte Linienprofil der relativen Fluoreszenzeinheiten über die Arterienwand vom Lumen bis zur Adventitia. Zusammenfassende Analyse des ASIC1-Ausdrucks in entweder (C) EC oder (D) SM von Lungenarterien aus Lungenschnitten. n = 5 Tiere pro Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen verglichen mit Šídáks multiplen Vergleichstests. (E) PCR der Schwanz-DNA, die Cre-vermittelte LoxP-Rekombination und Exzision von gezieltem Asic1a in derselben MHC CreER - Asic1a fl/fl -Maus vor (-) und nach (+) Tamoxifen (TAM)-Behandlung zeigt. (F) Asic1a- mRNA-Expression in Hirngewebe und intrapulmonalen Arterien (PA) in MHC CreER - Asic1a fl/fl- Mäusen mit Vehikel (-) oder TAM (+). Gescannte Bilder der Blots wurden invertiert und hinsichtlich Helligkeit/Kontrast angepasst. Um die Rolle von PAEC und PASMC ASIC1a bei CH-induzierter pulmonaler Hypertonie zu bestimmen, haben wir drei verschiedene CH-Behandlungsparadigmen entwickelt, die in
Abbildung 4A
dargestellt sind : 1) Vehikel: Asic1a +/+ , Asic1a −/- , Tek Cre – Asic1a fl/fl und MHC CreER – Asic1a fl/fl Mäuse wurden mit Vehikel (Maisöl) behandelt und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 2) vorbeugend (pTAM): Asic1a fl/fl (pTAM) und MHC CreER – Asic1a fl/fl (pTAM) Mäuse wurden mit TAM behandelt (5 Tage) und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 3) therapeutisch (tTAM): MHC CreER – Asic1a fl/fl (tTAM) Mäuse wurden 3 Wochen lang einer Kontrollgruppe oder CH ausgesetzt, um pulmonale Hypertonie festzustellen. Nach 3 Wochen CH wurden die Mäuse mit TAM behandelt (5 Tage mit con/CH-Exposition) und dann weitere 2 Wochen mit con/CH behandelt.
Tabelle 3
zeigt, dass die selektive Deletion von SM- oder EC- Asic1a den mittleren arteriellen Blutdruck oder die Herzfrequenz bei wachen Mäusen nicht signifikant veränderte und dem ähnelt, was wir zuvor bei Wildtyp-Mäusen beobachtet haben (
Detweiler et al., 2019
). Figur 4
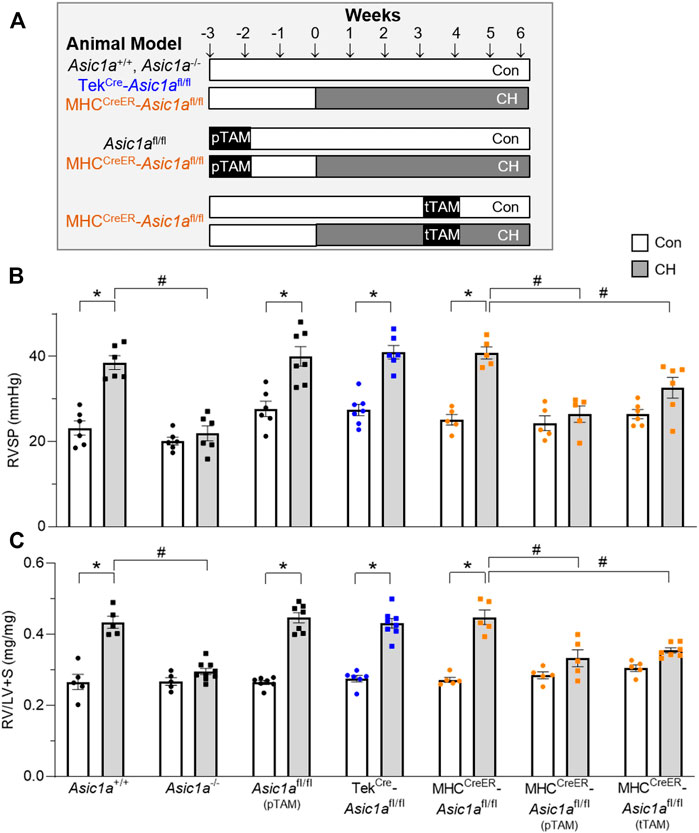 ABBILDUNG 4. SM-spezifischer Knockout von Asic1a schützt vor der Entwicklung von pulmonaler Hypertonie und kehrt eine bestehende hypoxische pulmonale Hypertonie um. (A) Experimentelles Design, das Behandlungen ohne TAM, präventive (pTAM, verabreicht vor CH) und therapeutische (tTAM, verabreicht nach bestehender pulmonaler Hypertonie) Behandlungen zeigt. ( Rechtsventrikulärer systolischer Druck (RVSP, mmHg) und (C) Fulton-Index (RV/LV + S) bei Asic1a +/+ , Asic1a −/- , Asic1a fl/fl (pTAM) , TekCre- Asic1a fl/fl , MHC CreER - Asic1a fl/fl , MHC CreER - Asic1a fl/fl (pTAM) oder MHC CreER - Asic1a fl/fl (tTAM) Mäusen unter Kontrollbedingungen (weiße Balken, Kreise) oder nach 6 Wochen CH (graue Balken, Quadrate). SM- Asic1a -Knockout wurde durch eine Behandlung mit Tamoxifen als vorbeugender (pTAM; vor Exposition gegenüber CH) oder therapeutischer Ansatz (tTAM; nach Auftreten von CH-induzierter pulmonaler Hypertonie) herbeigeführt. n = 5-8/Gruppe; analysiert durch zweiseitige ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p < 0,0001 für RVSP und RV/LV + S) wurden mit Šídáks Mehrfachvergleichstests untersucht; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. jeweilige genetische Kontrolle. Tisch 3
ABBILDUNG 4. SM-spezifischer Knockout von Asic1a schützt vor der Entwicklung von pulmonaler Hypertonie und kehrt eine bestehende hypoxische pulmonale Hypertonie um. (A) Experimentelles Design, das Behandlungen ohne TAM, präventive (pTAM, verabreicht vor CH) und therapeutische (tTAM, verabreicht nach bestehender pulmonaler Hypertonie) Behandlungen zeigt. ( Rechtsventrikulärer systolischer Druck (RVSP, mmHg) und (C) Fulton-Index (RV/LV + S) bei Asic1a +/+ , Asic1a −/- , Asic1a fl/fl (pTAM) , TekCre- Asic1a fl/fl , MHC CreER - Asic1a fl/fl , MHC CreER - Asic1a fl/fl (pTAM) oder MHC CreER - Asic1a fl/fl (tTAM) Mäusen unter Kontrollbedingungen (weiße Balken, Kreise) oder nach 6 Wochen CH (graue Balken, Quadrate). SM- Asic1a -Knockout wurde durch eine Behandlung mit Tamoxifen als vorbeugender (pTAM; vor Exposition gegenüber CH) oder therapeutischer Ansatz (tTAM; nach Auftreten von CH-induzierter pulmonaler Hypertonie) herbeigeführt. n = 5-8/Gruppe; analysiert durch zweiseitige ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p < 0,0001 für RVSP und RV/LV + S) wurden mit Šídáks Mehrfachvergleichstests untersucht; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. jeweilige genetische Kontrolle. Tisch 3
 TABELLE 3. Mittlerer arterieller Blutdruck (MABP) und Herzfrequenz bei genetisch veränderten Mäusen, die mit oder ohne Tamoxifen (pTAM) behandelt wurden. Ähnlich wie in unseren früheren Berichten führte die Exposition gegenüber CH zu einem signifikanten Anstieg des rechtsventrikulären systolischen Drucks (RVSP;
Abbildung 4B
) und der Rechtsherzhypertrophie (
Abbildung 4C
) bei Asic1a +/+- , aber nicht bei Asic1a −/- Mäusen (
Nitta et al., 2014
). CH führte zu einem ähnlichen Anstieg von RVSP und RV-Hypertrophie bei Asic1a fl/fl (pTAM) , Tek Cre - Asic1a fl/ fl und MHC CreER - Asic1a fl/fl -Mäusen (
Abbildung 4
). Die Asic1a fl/fl (pTAM) -Mäuse dienen nicht nur als Kontrolle für Tek Cre - Asic1a fl/fl- und MHC CreER - Asic1a fl/ fl-Mäuse, sondern liefern auch den Beweis, dass TAM die Entwicklung von pulmonaler Hypertonie nicht beeinflusst.
Tabelle 4
zeigt, dass CH bei keinem der transgenen Tiere die Körpermasse oder Herzfrequenz beeinflusst. Allerdings ist die Zunahme von RVSP und RV-Hypertrophie bei Asic1a fl/fl (pTAM) -, Tek Cre - Asic1a fl/fl- und MHC CreER - Asic1a fl/fl -Mäusen mit einer stärkeren RV-Kontraktilität und Arbeitsbelastung verbunden, wie durch erhöhte dP/dt max- bzw. Druck-Zeit-Index angezeigt wird. Diese Daten legen nahe, dass die EC-spezifische Deletion von Asic1 nicht zu einer Zunahme von RVSP und RV-Hypertrophie nach CH beiträgt. Im Gegensatz dazu verhinderte die SM-spezifische Deletion von Asic1a in MHC CreER - Asic1a fl/fl (pTAM) -Mäusen durch TAM-induzierte Cre-Rekombinase vor CH jegliche durch CH induzierte Zunahme von RVSP, RV-Hypertrophie, dP/dt max- und Druck-Zeit-Index (
Abbildung 4
und
Tabelle 3
). Darüber hinaus kehrte die Behandlung von MHC CreER - Asic1a fl/fl (tTAM) mit TAM in Woche drei der 6-wöchigen CH-Exposition die Erhöhungen von RVSP, RV-Hypertrophie, dP/dt max und Druck-Zeit-Index auf ähnliche Werte wie bei Kontrollmäusen um (
Abbildung 4
und
Tabelle 3
). Diese Daten zeigen, dass die Löschung von SM-, aber nicht EC- Asic1a sowohl CH-induzierte pulmonale Hypertonie als auch RV-Hypertrophie und -Dysfunktion verhindert und umkehrt. Tabelle 4
TABELLE 3. Mittlerer arterieller Blutdruck (MABP) und Herzfrequenz bei genetisch veränderten Mäusen, die mit oder ohne Tamoxifen (pTAM) behandelt wurden. Ähnlich wie in unseren früheren Berichten führte die Exposition gegenüber CH zu einem signifikanten Anstieg des rechtsventrikulären systolischen Drucks (RVSP;
Abbildung 4B
) und der Rechtsherzhypertrophie (
Abbildung 4C
) bei Asic1a +/+- , aber nicht bei Asic1a −/- Mäusen (
Nitta et al., 2014
). CH führte zu einem ähnlichen Anstieg von RVSP und RV-Hypertrophie bei Asic1a fl/fl (pTAM) , Tek Cre - Asic1a fl/ fl und MHC CreER - Asic1a fl/fl -Mäusen (
Abbildung 4
). Die Asic1a fl/fl (pTAM) -Mäuse dienen nicht nur als Kontrolle für Tek Cre - Asic1a fl/fl- und MHC CreER - Asic1a fl/ fl-Mäuse, sondern liefern auch den Beweis, dass TAM die Entwicklung von pulmonaler Hypertonie nicht beeinflusst.
Tabelle 4
zeigt, dass CH bei keinem der transgenen Tiere die Körpermasse oder Herzfrequenz beeinflusst. Allerdings ist die Zunahme von RVSP und RV-Hypertrophie bei Asic1a fl/fl (pTAM) -, Tek Cre - Asic1a fl/fl- und MHC CreER - Asic1a fl/fl -Mäusen mit einer stärkeren RV-Kontraktilität und Arbeitsbelastung verbunden, wie durch erhöhte dP/dt max- bzw. Druck-Zeit-Index angezeigt wird. Diese Daten legen nahe, dass die EC-spezifische Deletion von Asic1 nicht zu einer Zunahme von RVSP und RV-Hypertrophie nach CH beiträgt. Im Gegensatz dazu verhinderte die SM-spezifische Deletion von Asic1a in MHC CreER - Asic1a fl/fl (pTAM) -Mäusen durch TAM-induzierte Cre-Rekombinase vor CH jegliche durch CH induzierte Zunahme von RVSP, RV-Hypertrophie, dP/dt max- und Druck-Zeit-Index (
Abbildung 4
und
Tabelle 3
). Darüber hinaus kehrte die Behandlung von MHC CreER - Asic1a fl/fl (tTAM) mit TAM in Woche drei der 6-wöchigen CH-Exposition die Erhöhungen von RVSP, RV-Hypertrophie, dP/dt max und Druck-Zeit-Index auf ähnliche Werte wie bei Kontrollmäusen um (
Abbildung 4
und
Tabelle 3
). Diese Daten zeigen, dass die Löschung von SM-, aber nicht EC- Asic1a sowohl CH-induzierte pulmonale Hypertonie als auch RV-Hypertrophie und -Dysfunktion verhindert und umkehrt. Tabelle 4
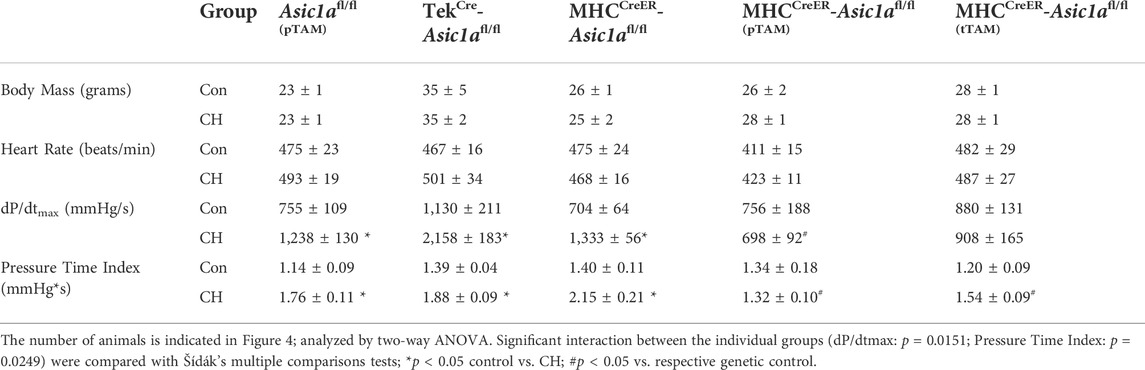 TABELLE 4. Körpermasse, Herzfrequenz, Herzkontraktilität und Druck-Zeit-Index bei anästhesierten Kontrollmäusen und genetisch veränderten CH-Mäusen, die mit oder ohne Tamoxifen (TAM) behandelt wurden. SM-spezifischer Knockout von Asic1a schwächt die Gefäßumgestaltung abDie Gefäßumgestaltung wurde mittels Immunfluoreszenz von SM α-Actin (SMA) in Gefäßen im Bereich von <25 μm, 25–50 μm und 50–100 μm beurteilt, wie in
Abbildung 5A gezeigt. CH verursachte bei Tek
Cre - Asic1a fl/fl- Mäusen eine signifikante Erhöhung der prozentualen Muskularisierung in jeder Arteriengröße . Einige Gefäße (∼5–10 %) von Tek Cre - Asic1a fl/fl- Mäusen, die CH ausgesetzt waren, wiesen hyperzelluläre Läsionen auf, die von den medialen und adventitialen Schichten nach außen in das angrenzende Lungenparenchym hineinragten (
Abbildung 5A
). Obwohl diese adventitiale Umgestaltung nicht als Teil der medialen Dicke analysiert wurde, wurde beobachtet, dass die Zellen in diesen Bereichen SMA mit einer geringeren Fluoreszenzintensität exprimierten als die mediale Schicht, was wahrscheinlich (Myo)Fibroblasten darstellt. Diese nach außen gerichtete Umgestaltung war im Wildtyp oder anderen transgenen Mausmodellen nicht vorhanden (
Nitta et al., 2014
;
Detweiler et al., 2019
;
Sheak et al., 2020
), was darauf hindeutet, dass die spezifische EC-Deletion von Asic1a die vaskuläre Umgestaltung erleichtern könnte. CH erhöhte den Prozentsatz der Muskularisierung bei MHC CreER - Asic1a fl/fl -Mäusen (keine TAM-induzierte Cre-Rekombinase), der sowohl durch pTAM- als auch durch tTAM-Behandlungen abgeschwächt wurde (
Abbildung 5B
). Die SM-spezifische Deletion von Asic1a hatte eine größere Wirkung bei der Verringerung der (Neo-)Muskularisierung in Arterien <25 µm, da es keinen signifikanten Unterschied im Vergleich zu Kontrollarterien gab. Darüber hinaus war die therapeutische Deletion von SM- Asic1a bei der Verringerung der arteriellen Muskularisierung wirksamer als die vorbeugende SM- Asic1a- Deletion. In 50–100 µm großen Arterien von MHC CreER - Asic1a fl/fl (tTAM) -Mäusen unterschied sich die Muskularisierung zwischen Kontroll- und CH-Mäusen nicht signifikant (
Abbildung 5B
). Abbildung 5
TABELLE 4. Körpermasse, Herzfrequenz, Herzkontraktilität und Druck-Zeit-Index bei anästhesierten Kontrollmäusen und genetisch veränderten CH-Mäusen, die mit oder ohne Tamoxifen (TAM) behandelt wurden. SM-spezifischer Knockout von Asic1a schwächt die Gefäßumgestaltung abDie Gefäßumgestaltung wurde mittels Immunfluoreszenz von SM α-Actin (SMA) in Gefäßen im Bereich von <25 μm, 25–50 μm und 50–100 μm beurteilt, wie in
Abbildung 5A gezeigt. CH verursachte bei Tek
Cre - Asic1a fl/fl- Mäusen eine signifikante Erhöhung der prozentualen Muskularisierung in jeder Arteriengröße . Einige Gefäße (∼5–10 %) von Tek Cre - Asic1a fl/fl- Mäusen, die CH ausgesetzt waren, wiesen hyperzelluläre Läsionen auf, die von den medialen und adventitialen Schichten nach außen in das angrenzende Lungenparenchym hineinragten (
Abbildung 5A
). Obwohl diese adventitiale Umgestaltung nicht als Teil der medialen Dicke analysiert wurde, wurde beobachtet, dass die Zellen in diesen Bereichen SMA mit einer geringeren Fluoreszenzintensität exprimierten als die mediale Schicht, was wahrscheinlich (Myo)Fibroblasten darstellt. Diese nach außen gerichtete Umgestaltung war im Wildtyp oder anderen transgenen Mausmodellen nicht vorhanden (
Nitta et al., 2014
;
Detweiler et al., 2019
;
Sheak et al., 2020
), was darauf hindeutet, dass die spezifische EC-Deletion von Asic1a die vaskuläre Umgestaltung erleichtern könnte. CH erhöhte den Prozentsatz der Muskularisierung bei MHC CreER - Asic1a fl/fl -Mäusen (keine TAM-induzierte Cre-Rekombinase), der sowohl durch pTAM- als auch durch tTAM-Behandlungen abgeschwächt wurde (
Abbildung 5B
). Die SM-spezifische Deletion von Asic1a hatte eine größere Wirkung bei der Verringerung der (Neo-)Muskularisierung in Arterien <25 µm, da es keinen signifikanten Unterschied im Vergleich zu Kontrollarterien gab. Darüber hinaus war die therapeutische Deletion von SM- Asic1a bei der Verringerung der arteriellen Muskularisierung wirksamer als die vorbeugende SM- Asic1a- Deletion. In 50–100 µm großen Arterien von MHC CreER - Asic1a fl/fl (tTAM) -Mäusen unterschied sich die Muskularisierung zwischen Kontroll- und CH-Mäusen nicht signifikant (
Abbildung 5B
). Abbildung 5
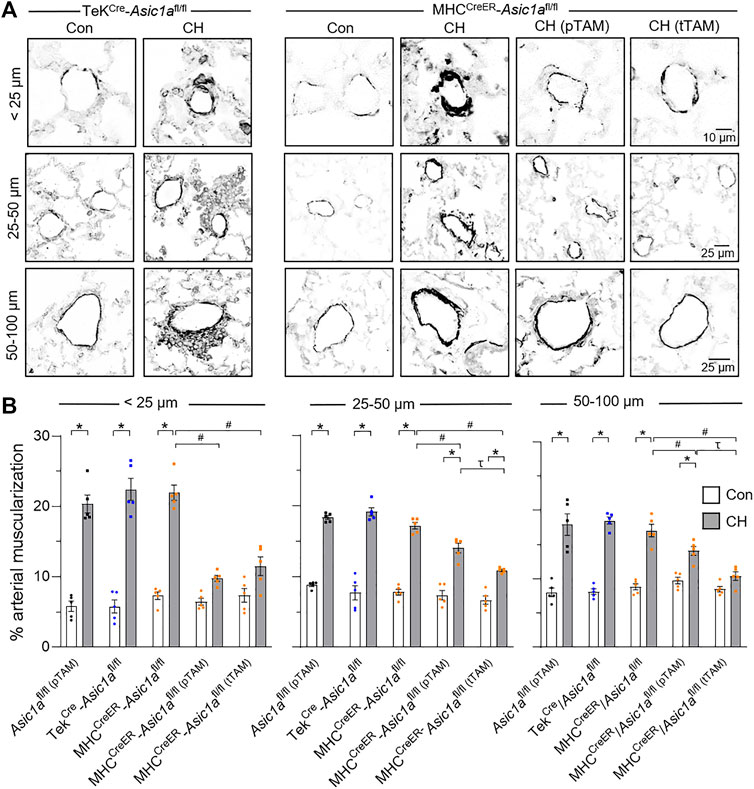 ABBILDUNG 5. SM ASIC1a trägt zur Gefäßumgestaltung nach CH bei. (A) Repräsentative SMA-Immunfluoreszenzbilder (schwarz) von kleinen Lungenarterien in Lungenschnitten von TekCre- Asic1a fl/fl- , MHC CreER - Asic1a fl/fl- , MHC CreER - Asic1a fl/fl- (pTAM-) oder MHC CreER - Asic1a fl/fl- (tTAM-) Mäusen unter Kontrollbedingungen (weiße Balken) oder nach 6 Wochen CH (gefüllte Balken). Fluoreszenzbilder wurden digital invertiert, um einen besseren Kontrast und eine bessere Sichtbarkeit der Immunfluoreszenz zu bieten. ( Prozentuale Muskulaturisierung berechnet als Prozent SMA-Grenzwertfläche geteilt durch Gesamtarterienfläche basierend auf Arteriendurchmesser: <25 μm (n = ∼30 Gefäße von vier Tieren/Gruppe), 25–50 μm (n = ∼100 Gefäße von vier Tieren/Gruppe) oder 50–100 μm (n = ∼50 Gefäße von vier Tieren/Gruppe); analysiert mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für alle Gefäßdurchmesserbereiche) verglichen mit Šídáks Mehrfachvergleichstests. * p < 0,05 vs. Kontrolle; # p < 0,05 vs. (-) TAM; und τ p < 0,05 pTAM vs. tTAM. Die Proliferation von PAECs und PASMCs in MHC CreER - Asic1a fl/fl -Mäusen wurde zusätzlich mittels Immunfluoreszenz ausgewertet, um Ki-67-positive Zellen zu identifizieren (
Abbildung 6
). Die Deletion von SM- Asic1a hatte keinen signifikanten Einfluss auf den Prozentsatz positiver Ki-67-Kerne in PAECs, reduzierte jedoch den Prozentsatz proliferierender PASMCs signifikant (
Abbildung 6B
). Zusammen zeigen diese Daten, dass SM- Asic1a zur CH-induzierten Gefäßumgestaltung beiträgt, indem es sowohl zur Muskulaturisierung als auch zur PASMC-Proliferation beiträgt. Abbildung 6
ABBILDUNG 5. SM ASIC1a trägt zur Gefäßumgestaltung nach CH bei. (A) Repräsentative SMA-Immunfluoreszenzbilder (schwarz) von kleinen Lungenarterien in Lungenschnitten von TekCre- Asic1a fl/fl- , MHC CreER - Asic1a fl/fl- , MHC CreER - Asic1a fl/fl- (pTAM-) oder MHC CreER - Asic1a fl/fl- (tTAM-) Mäusen unter Kontrollbedingungen (weiße Balken) oder nach 6 Wochen CH (gefüllte Balken). Fluoreszenzbilder wurden digital invertiert, um einen besseren Kontrast und eine bessere Sichtbarkeit der Immunfluoreszenz zu bieten. ( Prozentuale Muskulaturisierung berechnet als Prozent SMA-Grenzwertfläche geteilt durch Gesamtarterienfläche basierend auf Arteriendurchmesser: <25 μm (n = ∼30 Gefäße von vier Tieren/Gruppe), 25–50 μm (n = ∼100 Gefäße von vier Tieren/Gruppe) oder 50–100 μm (n = ∼50 Gefäße von vier Tieren/Gruppe); analysiert mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für alle Gefäßdurchmesserbereiche) verglichen mit Šídáks Mehrfachvergleichstests. * p < 0,05 vs. Kontrolle; # p < 0,05 vs. (-) TAM; und τ p < 0,05 pTAM vs. tTAM. Die Proliferation von PAECs und PASMCs in MHC CreER - Asic1a fl/fl -Mäusen wurde zusätzlich mittels Immunfluoreszenz ausgewertet, um Ki-67-positive Zellen zu identifizieren (
Abbildung 6
). Die Deletion von SM- Asic1a hatte keinen signifikanten Einfluss auf den Prozentsatz positiver Ki-67-Kerne in PAECs, reduzierte jedoch den Prozentsatz proliferierender PASMCs signifikant (
Abbildung 6B
). Zusammen zeigen diese Daten, dass SM- Asic1a zur CH-induzierten Gefäßumgestaltung beiträgt, indem es sowohl zur Muskulaturisierung als auch zur PASMC-Proliferation beiträgt. Abbildung 6
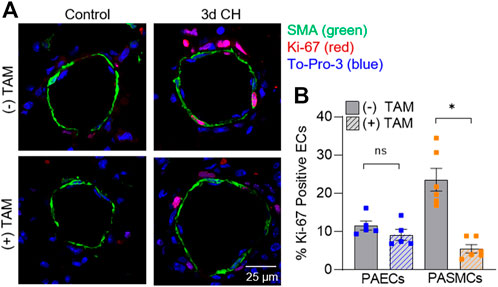 ABBILDUNG 6. SM ASIC1a trägt zur CH-induzierten PASMC-Proliferation bei. (A) Repräsentative Immunfluoreszenzbilder, die Ki-67 (rot), α-SMA (grün) und Sytox (blau) in kleinen Lungenarterien (<100 µm) von Kontrollmäusen und 3 Tage alten CH MHC CreER - Asic1a fl/fl -Mäusen nach Behandlung mit Vehikel oder Tamoxifen (TAM) zeigen. ( Zusammenfassungsdaten für den Prozentsatz Ki-67-positiver Zellen in Endothelzellen (EC, blau) und glatten Muskelzellen (SMC, orange). Zusammenfassungsdaten für Kontrollmäuse werden nicht angezeigt, da keine Ki-67-positiven Zellen nachweisbar waren. n = 75 Gefäße von fünf Tieren (je 15 Gefäße) pro Gruppe; analysiert mit ungepaartem t -Test; # p < 0,05 vs. (-) TAM 3 Tage CH; ns = nicht signifikant. ASIC1 trägt zur Migration und Verbreitung von PASMC beiPASMCs wurden in vitro einer Hypoxie ausgesetzt , anschließend wurden Migration und Proliferation mittels Transwell-Assays bzw. Durchflusszytometrie für BrdU-positive Zellen untersucht. Hypoxie erhöhte den Prozentsatz migrierender mPASMCs von Asic1a +/+ -Mäusen signifikant , nicht jedoch von Asic1a −/- -Mäusen (
Abbildungen 7A, B ). Die Proliferation, gemessen anhand des BrdU-Einbaus, war bei mPASMCs von
Asic1a −/- -Mäusen unter Normoxie und nach Exposition gegenüber 24 und 48 h Hypoxie signifikant geringer (
Abbildung 7C
). Abbildung 7
ABBILDUNG 6. SM ASIC1a trägt zur CH-induzierten PASMC-Proliferation bei. (A) Repräsentative Immunfluoreszenzbilder, die Ki-67 (rot), α-SMA (grün) und Sytox (blau) in kleinen Lungenarterien (<100 µm) von Kontrollmäusen und 3 Tage alten CH MHC CreER - Asic1a fl/fl -Mäusen nach Behandlung mit Vehikel oder Tamoxifen (TAM) zeigen. ( Zusammenfassungsdaten für den Prozentsatz Ki-67-positiver Zellen in Endothelzellen (EC, blau) und glatten Muskelzellen (SMC, orange). Zusammenfassungsdaten für Kontrollmäuse werden nicht angezeigt, da keine Ki-67-positiven Zellen nachweisbar waren. n = 75 Gefäße von fünf Tieren (je 15 Gefäße) pro Gruppe; analysiert mit ungepaartem t -Test; # p < 0,05 vs. (-) TAM 3 Tage CH; ns = nicht signifikant. ASIC1 trägt zur Migration und Verbreitung von PASMC beiPASMCs wurden in vitro einer Hypoxie ausgesetzt , anschließend wurden Migration und Proliferation mittels Transwell-Assays bzw. Durchflusszytometrie für BrdU-positive Zellen untersucht. Hypoxie erhöhte den Prozentsatz migrierender mPASMCs von Asic1a +/+ -Mäusen signifikant , nicht jedoch von Asic1a −/- -Mäusen (
Abbildungen 7A, B ). Die Proliferation, gemessen anhand des BrdU-Einbaus, war bei mPASMCs von
Asic1a −/- -Mäusen unter Normoxie und nach Exposition gegenüber 24 und 48 h Hypoxie signifikant geringer (
Abbildung 7C
). Abbildung 7
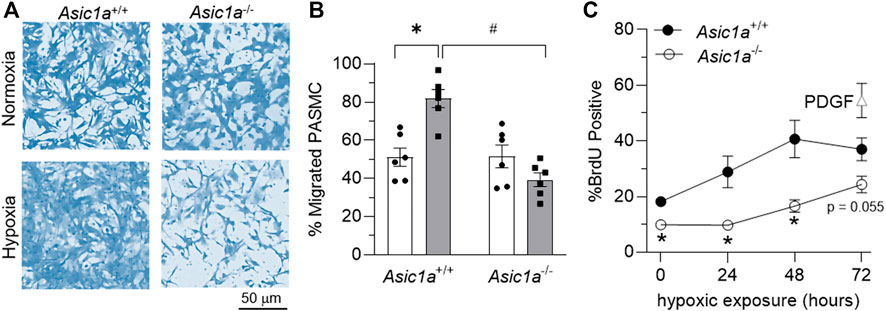 ABBILDUNG 7. ASIC1a trägt zur Migration und Proliferation von mPASMC bei. (A) Repräsentative Hellfeldbilder von Coomassie-gefärbten mPASMC und ( zusammenfassende Daten für % migrierter mPASMC von Asic1a +/+ und Asic1a −/- Mäusen, die Normoxie (5 % CO2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO2 , 2 % O2 ; blaue Quadrate) für 24 hn = 6 Tiere/Gruppe ausgesetzt waren; * p < 0,05 vs. Hypoxie und # p < 0,05 Asic1a +/+ ; analysiert durch zweiseitige ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0003) verglichen unter Verwendung von Šídáks Mehrfachvergleichstests. (C) Durchflusszytometrieanalyse des Prozentsatzes an mPASMC von Asic1a +/+ und Asic1a −/- Mäusen mit BrdU-Einbau unter Normoxie (0 h; 5 % CO 2 , 95 % Luft) oder Hypoxie (5 % CO 2 , 2 % O 2 ) für 24, 48 oder 72 h. Normoxische mPASMC von Asic1a +/+ wurden 72 h lang mit PDGF-BB (20 ng/ml) als positive Kontrolle inkubiert. n = fünf bis acht Tiere/Gruppe, zu jedem Zeitpunkt durch ungepaarte t-Tests analysiert, * p < 0,05. Um festzustellen, ob diese Erkenntnisse auf menschliche PASMCs (hPASMCs) übertragbar sind, haben wir die Auswirkungen von Hypoxie auf ASIC1-Expression, -Migration und -Proliferation in hPASMCs in Anwesenheit oder Abwesenheit einer ASIC1-Hemmung untersucht. Obwohl 12-stündige Hypoxie die Gesamtexpression von ASIC1 nicht veränderte (
Abbildung 8A, B
), vergrößerte sie die Plasmamembran (Zelloberfläche) im Vergleich zur zytosolischen Expression signifikant (
Abbildung 8C
). Dies steht im Einklang mit früheren Studien an 4 Wochen CH-exponierten Ratten, bei denen die Gesamtexpression von ASIC1 unverändert blieb, Hypoxie jedoch eine subzelluläre Translokation von ASIC1 zur Plasmamembran verursachte (
Herbert et al., 2016
,
2018
). Der Prozentsatz der Reinvasion von hPASMCs war nach 12-stündiger Hypoxie höher als nach Normoxie und wurde durch eine Vorbehandlung mit Amilorid oder PcTX1 verhindert (
Abbildung 8D, E
). Die durch Hypoxie induzierte Proliferation wurde in hPASMCs zusätzlich durch Amilorid und PcTX1 blockiert (
Abbildung 8F
). Zusammengenommen deuten diese Daten auf eine Beteiligung von ASIC1 an der durch Hypoxie vermittelten Proliferation und Migration von hPASMCs hin. Abbildung 8
ABBILDUNG 7. ASIC1a trägt zur Migration und Proliferation von mPASMC bei. (A) Repräsentative Hellfeldbilder von Coomassie-gefärbten mPASMC und ( zusammenfassende Daten für % migrierter mPASMC von Asic1a +/+ und Asic1a −/- Mäusen, die Normoxie (5 % CO2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO2 , 2 % O2 ; blaue Quadrate) für 24 hn = 6 Tiere/Gruppe ausgesetzt waren; * p < 0,05 vs. Hypoxie und # p < 0,05 Asic1a +/+ ; analysiert durch zweiseitige ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0003) verglichen unter Verwendung von Šídáks Mehrfachvergleichstests. (C) Durchflusszytometrieanalyse des Prozentsatzes an mPASMC von Asic1a +/+ und Asic1a −/- Mäusen mit BrdU-Einbau unter Normoxie (0 h; 5 % CO 2 , 95 % Luft) oder Hypoxie (5 % CO 2 , 2 % O 2 ) für 24, 48 oder 72 h. Normoxische mPASMC von Asic1a +/+ wurden 72 h lang mit PDGF-BB (20 ng/ml) als positive Kontrolle inkubiert. n = fünf bis acht Tiere/Gruppe, zu jedem Zeitpunkt durch ungepaarte t-Tests analysiert, * p < 0,05. Um festzustellen, ob diese Erkenntnisse auf menschliche PASMCs (hPASMCs) übertragbar sind, haben wir die Auswirkungen von Hypoxie auf ASIC1-Expression, -Migration und -Proliferation in hPASMCs in Anwesenheit oder Abwesenheit einer ASIC1-Hemmung untersucht. Obwohl 12-stündige Hypoxie die Gesamtexpression von ASIC1 nicht veränderte (
Abbildung 8A, B
), vergrößerte sie die Plasmamembran (Zelloberfläche) im Vergleich zur zytosolischen Expression signifikant (
Abbildung 8C
). Dies steht im Einklang mit früheren Studien an 4 Wochen CH-exponierten Ratten, bei denen die Gesamtexpression von ASIC1 unverändert blieb, Hypoxie jedoch eine subzelluläre Translokation von ASIC1 zur Plasmamembran verursachte (
Herbert et al., 2016
,
2018
). Der Prozentsatz der Reinvasion von hPASMCs war nach 12-stündiger Hypoxie höher als nach Normoxie und wurde durch eine Vorbehandlung mit Amilorid oder PcTX1 verhindert (
Abbildung 8D, E
). Die durch Hypoxie induzierte Proliferation wurde in hPASMCs zusätzlich durch Amilorid und PcTX1 blockiert (
Abbildung 8F
). Zusammengenommen deuten diese Daten auf eine Beteiligung von ASIC1 an der durch Hypoxie vermittelten Proliferation und Migration von hPASMCs hin. Abbildung 8
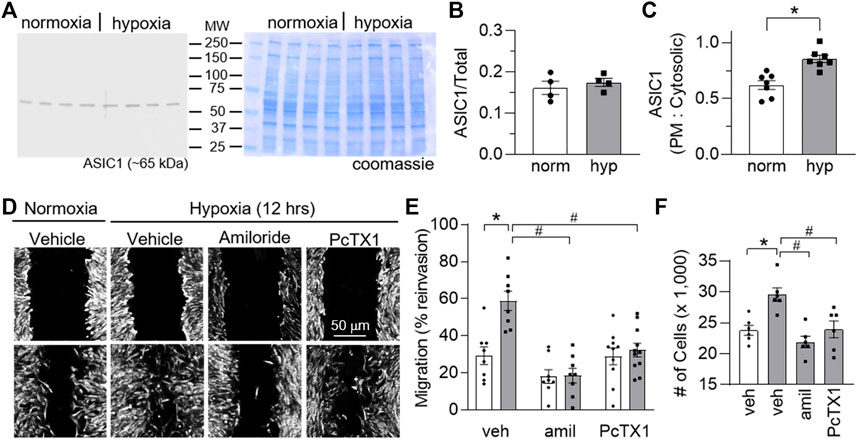 ABBILDUNG 8. ASIC1 trägt zur Migration und Proliferation menschlicher PASMC bei. (A) Repräsentative Western Blots von ASIC1 (vorhergesagt ∼65 kDa) und entsprechender Coomassie-gefärbter Blot und ( Zusammenfassung der Daten, die die ASIC1-Expression (normalisiert auf die gesamte Spur des Coomassie-gefärbten Blots) in menschlichen PASMC (hPASMC) zeigen, die Normoxie (Norm, 5 % CO2 , 95 % Luft; weiße Balken/Kreise) oder 12 Stunden Hypoxie (Hyp, 5 % CO2 , 2 % O2 ; graue Balken/Quadrate) ausgesetzt waren. Die gescannten Bilder des Films wurden in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. (C) Zusammenfassung der Daten, die die Western-Blot-Analyse der Expression von biotinyliertem (Plasmamembran-) und zytosolischem ASIC1-Protein in hPASMC zeigen, die Normoxie oder Hypoxie ausgesetzt waren. (D) Hellfeldbilder von hPASMC unmittelbar nach dem Kratzer (Basiszeit 0; obere Bildreihe) oder 12 Stunden nach der Exposition (untere Bildreihe) gegenüber Normoxie (5 % CO 2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO 2 , 2 % O 2 ; blaue Quadrate) und (E) zusammenfassende Daten, die die prozentuale Reinvasion von hPASMC in den verletzten Bereich in Gegenwart von Vehikel, Amilorid oder PcTX1 zeigen. n = 8–11/Gruppe; analysiert mittels zweiseitiger ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p = 0,0032) wurden mithilfe von Šídáks Mehrfachvergleichstests verglichen. (F) Zunahme der Zellzahl
ABBILDUNG 8. ASIC1 trägt zur Migration und Proliferation menschlicher PASMC bei. (A) Repräsentative Western Blots von ASIC1 (vorhergesagt ∼65 kDa) und entsprechender Coomassie-gefärbter Blot und ( Zusammenfassung der Daten, die die ASIC1-Expression (normalisiert auf die gesamte Spur des Coomassie-gefärbten Blots) in menschlichen PASMC (hPASMC) zeigen, die Normoxie (Norm, 5 % CO2 , 95 % Luft; weiße Balken/Kreise) oder 12 Stunden Hypoxie (Hyp, 5 % CO2 , 2 % O2 ; graue Balken/Quadrate) ausgesetzt waren. Die gescannten Bilder des Films wurden in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. (C) Zusammenfassung der Daten, die die Western-Blot-Analyse der Expression von biotinyliertem (Plasmamembran-) und zytosolischem ASIC1-Protein in hPASMC zeigen, die Normoxie oder Hypoxie ausgesetzt waren. (D) Hellfeldbilder von hPASMC unmittelbar nach dem Kratzer (Basiszeit 0; obere Bildreihe) oder 12 Stunden nach der Exposition (untere Bildreihe) gegenüber Normoxie (5 % CO 2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO 2 , 2 % O 2 ; blaue Quadrate) und (E) zusammenfassende Daten, die die prozentuale Reinvasion von hPASMC in den verletzten Bereich in Gegenwart von Vehikel, Amilorid oder PcTX1 zeigen. n = 8–11/Gruppe; analysiert mittels zweiseitiger ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p = 0,0032) wurden mithilfe von Šídáks Mehrfachvergleichstests verglichen. (F) Zunahme der Zellzahl
www.frontiersin.org/journals/molecular-b...olb.2022.989809/full
Der säureempfindliche Ionenkanal 1a (ASIC1a) ist ein spannungsunabhängiger, nicht selektiver Kationenkanal, der sowohl Na + als auch Ca2 + leitet . Die Aktivierung von ASIC1a löst eine Depolarisation der Plasmamembran aus und stimuliert intrazelluläre Ca2 + -abhängige Signalwege in mehreren Zelltypen, darunter Gefäßglattmuskelzellen (SM) und Endothelzellen (ECs). Frühere Studien haben gezeigt, dass ein Anstieg des pulmonalen Gefäßwiderstands, der mit chronischer Hypoxie (CH) induzierter pulmonaler Hypertonie einhergeht, ASIC1a erfordert, um eine verstärkte pulmonale Vasokonstriktion und Gefäßumgestaltung hervorzurufen. Sowohl SM- als auch EC-Dysfunktion treiben diese Prozesse an; die Beteiligung von ASIC1a an diesen verschiedenen Zelltypen ist jedoch unbekannt. Mithilfe des Cre-LoxP-Systems zur Erzeugung zelltypspezifischer Asic1a -Knockout-Mäuse testeten wir die Hypothese, dass SM- Asic1a zu CH-induzierter pulmonaler Hypertonie und Gefäßumbau beiträgt, während EC- Asic1a der Entwicklung von CH-induzierter pulmonaler Hypertonie entgegenwirkt. Der Schweregrad der pulmonalen Hypertonie blieb bei Mäusen mit spezifischer Deletion von EC- Asic1a (Tek Cre - Asic1a fl/fl ) unverändert. Ähnlich wie globale Asic1a- Knockout-Mäuse ( Asic1a −/- ) waren Mäuse mit spezifischer Deletion von SM- Asic1a (MHC CreER - Asic1a fl/fl ) jedoch vor der Entwicklung von CH-induzierter pulmonaler Hypertonie und Rechtsherzhypertrophie geschützt. Darüber hinaus war die pulmonale Hypertonie rückgängig gemacht worden, als die Deletion von SM- Asic1a bei konditionellen MHC CreER - Asic1a fl/fl- Mäusen mit bestehender pulmonaler Hypertonie eingeleitet wurde. Die durch CH induzierte Gefäßumgestaltung war auch in den Lungenarterien von MHC CreER - Asic1a fl/fl- Mäusen deutlich abgeschwächt. Diese Ergebnisse wurden zusätzlich durch eine verringerte, durch CH induzierte Proliferation und Migration von pulmonalarteriellen glatten Muskelzellen (PASMCs) von Asic1a −/- Mäusen unterstützt. Zusammen zeigen diese Daten, dass SM-, aber nicht EC- Asic1a zu durch CH induzierter pulmonaler Hypertonie und Gefäßumgestaltung beiträgt. Darüber hinaus liefern diese Studien Beweise für das therapeutische Potenzial der ASIC1a-Hemmung zur Umkehrung der pulmonalen Hypertonie. 1. EinleitungUnter normalen physiologischen Bedingungen wird der Lungenkreislauf in einem Zustand mit niedrigem Druck und geringem Widerstand gehalten, mit geringem oder keinem Ruhegefäßtonus. Unter pathologischen Bedingungen führt ein anhaltender Anstieg des pulmonalen Gefäßwiderstands zur Entwicklung einer pulmonalen Hypertonie. Pulmonale Hypertonie ist eine fortschreitende und häufig tödlich verlaufende Lungengefäßerkrankung, die durch einen mittleren pulmonalarteriellen Druck von >20 mmHg definiert ist ( Simonneau et al., 2019 ). Mit der Zeit erhöhen der erhöhte Gefäßwiderstand und der pulmonalarterielle Druck die Nachlast des rechten Ventrikels. Wenn die Anpassungsmechanismen der Dilatation und Hypertrophie des rechten Ventrikels den hohen Gefäßwiderstand in der Lunge nicht mehr kompensieren können, tritt eine Rechtsherzinsuffizienz auf, die mit einer schlechten Prognose verbunden ist.Obwohl pulmonale Hypertonie verschiedene Ursachen haben kann, kann der Anstieg des pulmonalen Gefäßwiderstands bei allen Formen der pulmonalen Hypertonie auf eine Kombination aus anhaltender pulmonaler Vasokonstriktion und Gefäßumbau zurückgeführt werden. Eine verstärkte Vasokonstriktion ist mit einer Funktionsstörung der pulmonalarteriellen Endothelzellen (PAEC) und einer Hyperreaktivität der pulmonalarteriellen glatten Muskelzellen (PASMCs) verbunden ( Budhiraja et al., 2004 ; Lin et al., 2004 ; Nagaoka et al., 2004 ; Jernigan et al., 2008 ; Broughton et al., 2010 ; Weise-Cross et al., 2018 ). Die Auswirkungen der Umgestaltung auf den pulmonalvaskulären Widerstand sind vor allem auf die Verdickung der Intima und/oder Mediaschicht kleiner Muskelarterien und die distale Neomuskularisierung zurückzuführen, die sich durch das Auftreten von Zellen äußert, die glatte Muskelzellen (SM)-spezifische Marker in normalerweise nicht-muskulären präkapillären, intraazinären Gefäßen exprimieren. Diese komplexe Pathogenese wird vermutlich durch eine Verletzung und Apoptose von Endothelzellen (EC) eingeleitet, gefolgt von der Entstehung übermäßiger Proliferation und Migration apoptoseresistenter PAECs und PASMCs sowie zellulärer Transdifferenzierung in Form eines EC-mesenchymalen Übergangs und phänotypischer SM-Transformationen ( Voelkel & Tuder, 2000 ; Shimoda & Laurie, 2013 ; Gao et al., 2016 ). Stoffwechselstörungen, die die aerobe Glykolyse fördern und die oxidative Atmung der Mitochondrien hemmen, führen nachweislich sowohl bei Tiermodellen als auch bei Patienten mit pulmonaler Hypertonie zu umfassenden Umbauten des rechten Ventrikels und der Gefäße ( McMurtry et al., 2004 ; Bonnet et al., 2006 ; Xu et al., 2007 ; Sutendra et al., 2010 ; Fessel et al., 2012 ; Dromparis et al., 2013 ; Pak et al., 2013 ). Die Umstellung des Zellstoffwechsels auf Milchsäuregärung führt zu einem pathologischen Anstieg des extrazellulären Säuregehalts. Mehrere Ionenkanäle werden entweder direkt gesteuert oder ihre Aktivität wird durch Änderungen des intra- und extrazellulären pH-Werts moduliert. Dazu gehören die säureempfindlichen Ionenkanäle (ASIC), der transiente Rezeptorpotential-Vanilloid-Rezeptor 1 (TRPV1), der transiente Rezeptorpotential-Ankyrin-Repeat-Rezeptor 1 (TRPA1), einige Zwei-Poren-Domänen-Kanäle (K2P), nach innen gleichrichtende K + -Kanäle (Kir) sowie spannungsgesteuerte Na +- , Ca2 + - und K + -Kanäle ( Harguindey et al., 2017 ).Säureempfindliche Ionenkanäle (ASICs) stellen eine Unterfamilie der Amilorid-sensitiven Degenerin/Epithel-Na + -Kanäle (Deg/ENaC) dar, die H + -gesteuerte, spannungsunempfindliche Kationenkanäle bilden. Ähnlich wie ENaCs sind ASICs hochselektiv für Na + gegenüber anderen Ionen, mit Ausnahme von ASIC1a, das zusätzlich Ca2 + leitet ( Waldmann et al., 1997 ; Xiong et al., 2004 ; Yermolaieva et al., 2004 ). Der Einstrom von Na + und Ca2 + trägt zur Membrandepolarisation, Aktivierung von Ca2 + -Calmodulin-abhängigen Mechanismen und anderen Second-Messenger-Wegen bei, was die vielfältigen Rollen verdeutlicht, die ASIC1a bei der intrazellulären Signalgebung und Erregbarkeit unter normalen wie auch pathologischen Bedingungen spielt. ASICs wurden vor allem in Neuronen untersucht, da sie im gesamten zentralen und peripheren Nervensystem ubiquitär exprimiert werden. Folglich ist es weniger bekannt, dass ASICs in einer Vielzahl anderer Zelltypen exprimiert werden, darunter Oligodendrozyten, Mesenchym-, Epithel-, Endothel-, Muskel-, Fett-/Endokrin- und Immunzellen, wo sie mit einer Reihe von Pathologien in Verbindung gebracht wurden ( Foster et al., 2021 ; Karlsson et al., 2021 ). Obwohl die Expression von ASIC1 in vaskulären SM und ECs berichtet wurde ( Grifoni et al., 2008 ; Jernigan et al., 2009 ; Chung et al., 2010 ; Akanji et al., 2019 ; Garcia et al., 2020 ; Redd et al., 2021 ), ist über die funktionelle Rolle von ASIC1 bei der Regulierung der vaskulären Homöostase bei Krankheitszuständen weniger bekannt.Frühere Studien in unserem Labor haben eine neue Rolle von ASIC1a bei der Entwicklung einer durch chronische Hypoxie (CH) induzierten pulmonalen Hypertonie identifiziert ( Nitta et al., 2014 ). ASIC1 wird sowohl in PASMCs als auch in PAECs exprimiert. Der Beitrag von ASIC1a zu den pathologischen Mechanismen, die zur Umgestaltung der Pulmonalarterien und zur Entwicklung von pulmonaler Hypertonie in diesen beiden Gefäßzelltypen führen, ist jedoch unklar. Während frühere Studien darauf hinweisen, dass PASMC ASIC1a eine pulmonale Vasokonstriktion vermittelt ( Jernigan et al., 2012 ), ist die funktionelle Rolle von ASIC1 in PAECs unbekannt. Basierend auf Studien, die zeigen, dass EC ASIC1 in Mesenterialarterien zur endothelialabhängigen Vasodilatation beiträgt ( Garcia et al., 2018 ), spekulieren wir, dass PAEC ASIC1a vor der Entwicklung von pulmonaler Hypertonie schützen könnte. Um die Hypothesen zu testen, dass SM- Asic1a zu CH-induzierter pulmonaler Hypertonie und vaskulärer Umgestaltung beiträgt und EC- Asic1a der Entwicklung von CH-induzierter pulmonaler Hypertonie entgegenwirkt, werden wir das Cre-loxP-System verwenden, um Mäuse mit selektiver EC- Asic1a- Deletion (Tek Cre - Asic1a fl/fl ) oder induzierbarer SM- Asic1a- Deletion (MHC CreER - Asic1a fl/fl ) zu erzeugen.Materialen und MethodenEthische GenehmigungAlle in dieser Studie verwendeten Protokolle wurden vom Institutional Animal Care and Use Committee der University of New Mexico School of Medicine (Protokoll Nr. 19-200899-HSC) geprüft und genehmigt und entsprechen den Richtlinien der National Institutes of Health für den Umgang mit Tieren. Alle Tiere wurden mit einer Überdosis Pentobarbital-Natrium (200 mg/kg, ip) betäubt und nach dem Bewusstseinsverlust sofort durch Ausbluten getötet.TiereDie Studien wurden an erwachsenen männlichen Wildtyp-Mäusen ( Asic1a +/+ ) oder verschiedenen transgenen Mäusen (12–16 Wochen alt) durchgeführt, wie in Tabelle 1 gezeigt. Um Asic1a in ECs oder SM selektiv zu löschen , wurden Asic1a fl/fl -Mäuse mit Tek Cre- bzw. MHC CreER -transgenen Mäusen gekreuzt. Es wurden homozygote und/oder heterozygote Mäuse gezüchtet und die Cre-Transgenexpression sowie die Deletion des Asic1a -Gens wurden durch PCR und Agarosegelelektrophorese bestätigt ( Tabelle 1 ). Die Tiere wurden eins bis fünf pro Käfig in einer speziellen pathogenfreien (SPF) Tierpflegeeinrichtung untergebracht und bei einem Hell-Dunkel-Rhythmus von 12:12 Stunden gehalten. Standardfutter (Teklad sojaproteinfreie Diät Nr. 2920, Envigo) und Wasser wurden ad libitum bereitgestellt . Die Tiere wurden zufällig den Versuchsgruppen zugeteilt und die Genotyp- und Behandlungszuweisungen wurden den Forschern wenn möglich nicht mitgeteilt. Es wurden ausschließlich männliche Mäuse untersucht, da die Expression von iCreER T2 unter der Kontrolle des SM-Promoters auf dem Y-Chromosom eingefügt ist. Darüber hinaus zeigten unsere bisherigen Daten keine signifikante Interaktion zwischen Geschlecht und der Entwicklung von hypoxischer pulmonaler Hypertonie ( Nitta et al., 2014 ). Zur Induktion der Cre-Aktivität wurden MHC CreER - Asic1a fl/fl -Mäusen an fünf aufeinanderfolgenden Tagen einmal täglich 75 mg/kg Tamoxifen (TAM; Sigma-Aldrich, CAS-Nr. 10540-29–1) in Maisöl injiziert. Nach 14 Tagen wurde die Cre-Rekombinase anhand des Schwanz-DNA-Primerpaars 5'-LoxP-Vorwärts- und 3'-LoxP-Rückwärtsprimerpaars bestimmt ( Tabelle 1 ). Tamoxifen wurde MHC CreER - Asic1a fl/fl -Mäusen verabreicht , um einen SM- Asic1a- Knockout zu induzieren, entweder 1) 2 Wochen vor CH als vorbeugendes (pTAM) Protokoll oder 2) nach 3 Wochen CH als therapeutisches (tTAM) Protokoll, um die Umkehrung einer bestehenden pulmonalen Hypertonie zu beurteilen. Wir haben zuvor gezeigt, dass Mäuse nach 3-wöchiger CH-Exposition pulmonale Hypertonie entwickeln ( Sheak et al., 2020 ). Die Asic1a- Disruptierung wurde anhand der Gesamt-RNA (1 µg) im Gehirn (positive Kontrolle) und isolierten Lungenarterien mittels RT-PCR beurteilt. Die Gesamt-RNA wurde mit TRIzol extrahiert und in cDNA rücktranskribiert (Transcription First-Strand cDNA Synthesis Kit, Roche, 04379012001). Die Amplifikation von Asic1a erfolgte mittels PCR (iCycler, Bio-Rad) unter Verwendung von REDExtract-N_Amp PCR Ready Mix (Sigma-Aldrich, XNAT) und Asic1Primer: vorwärts: 5′ CACATGCCAGGGGATGCCCC 3′ und rückwärts: 5′ AGCCGGTGCTTAATGACCTC 3‘ (410 bp). Das PCR-Produkt wurde mittels Gelelektrophorese auf einem 3%igen Agarosegel aufgetrennt und zur Visualisierung unter UV-Licht mit Ethidiumbromid gefärbt. Tabelle 1
zusammenfassende Daten, die den Prozentsatz Ki-67-positiver pulmonaler arterieller Endothelzellen (PAECs, blaue Kreise) und pulmonaler arterieller glatter Muskelzellen (PASMCs, orange Quadrate) unter Kontrollbedingungen oder nach Exposition gegenüber CH (3, 7 oder 28 Tage) zeigen. n = 3 Tiere pro Gruppe (für jedes Tier wurden im Durchschnitt ca. 20 Gefäße ermittelt); analysiert als einfaktorielle ANOVA für jeden Zelltyp und einzelne Gruppen verglichen mit Šídáks Mehrfachvergleichstests. Zusammenfassung der Daten, die den Prozentsatz Ki-67-positiver (C) PAECs und (D) PASMCs in kleinen Lungenarterien (<100 µm) von Asic1a +/+ und Asic1a −/- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition zeigen. n = fünf bis acht Tiere (durchschnittlich ∼15 Gefäße pro Tier); Analyse mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für PAECs und PASMCs) wurden mit Šídáks Mehrfachvergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. entsprechende 3-tägige CH; ns = nicht signifikant. Als nächstes ermittelten wir, ob die durch CH induzierte Zunahme der PASMC-Proliferation mit einem Verlust des kontraktilen Phänotyps verbunden ist, indem wir die Proteinexpression der SM-Myosin-Schwerkette (MHC) im Lungengewebe nach 0 (Con), 1, 2, 3, 5 und 7 Tagen CH analysierten (
Abbildung 2A
). In Übereinstimmung mit dem höchsten Prozentsatz Ki-67-positiver PASMCs nach 3 Tagen CH (
Abbildung 1B
) war MHC nach 3 Tagen CH bei Asic1a +/+- Tieren signifikant erniedrigt (
Abbildung 2B, C
). Nach 7 Tagen CH war MHC im Vergleich zu den Kontrollwerten erhöht (
Abbildung 2B
). CH verringerte die MHC-Spiegel bei Asic1a −/-- Mäusen nicht (
Abbildung 2C
). Hypoxie veränderte die Expressionsniveaus von GAPDH nicht ( p = 0,2082). Zusammengenommen deuten diese Daten darauf hin, dass ASIC1a zur Proliferation von PAEC und PASMC sowie zu phänotypischen Veränderungen von PASMC beiträgt, die in den Lungenarterien als Reaktion auf CH-Exposition auftreten. Figur 2
die Wirkung von CH-Exposition (1–7 Tage) auf die Expression des MHC-zu-GAPDH-Proteins in Gesamtlungenhomogenaten von Asic1a +/+- Tieren. Das gescannte Bild des Films wurde in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. n = 6/Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen wurden mit Šídáks mehrfachen Vergleichstests verglichen. (C) Zusammenfassende Daten für MHC zu GAPDH in Gesamtlungen von Asic1a +/+- und Asic1a −/-- Mäusen unter Kontrollbedingungen oder nach 3-tägiger CH-Exposition. n = 5-7 Tiere/Gruppe; analysiert mit zweifaktorieller ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0022) wurden mit Šídáks mehrfachen Vergleichstests verglichen; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. Asic1a +/+ Mäuse. SM-spezifisches Knockout von Asic1a schützt vor der Entwicklung von hypoxischer pulmonaler Hypertonie und kehrt diese umUm die spezifische Rolle von ASIC1a bei der Umgestaltung von PAEC und PASMC bei pulmonaler Hypertonie zu bestimmen, haben wir Mäuse mit entweder EC- (Tek Cre - Asic1a fl/fl ) oder bedingter SM-spezifischer (MHC CreER - Asic1a fl/fl ) Deletion von Asic1a erzeugt. Wie bereits zuvor bei Asic1a +/+ und Asic1a −/- Mäusen gezeigt (
Nitta et al., 2014 ), wurde ASIC1 als punktförmige Fluoreszenz in den PASMCs und PAECs von
Asic1a fl/fl und MHC CreER - Asic1a fl/fl (ohne TAM-induzierte Cre-Rekombinase) Mäusen nachgewiesen (
Abbildungen 3A,B
). Das Linienprofil durch die Gefäßwand zeigt, dass bei Tek Cre – Asic1a fl/fl- Mäusen die Expression von ASIC1 in ECs fehlt, die Expression in PASMCs jedoch erhalten bleibt, während bei MHC CreER – Asic1a fl/fl (TAM) (mit TAM-induzierter Cre-Rekombinase) die Expression von ASIC1 in PASMCs fehlt, die Expression in PAECs jedoch erhalten bleibt (
Abbildungen 3A–D
).
Abbildung 3E
zeigt die TAM-induzierte Cre-Rekombination zwischen loxP- Stellen und den Verlust der dazwischenliegenden Genomsequenz (Exons 2–3) in der Schwanz-DNA vor und nach TAM im selben Tier. ASIC1 wird im zentralen Nervensystem stark exprimiert und
Abbildung 3F zeigt, dass die TAM-induzierte Cre-Rekombinase
die Asic1a- mRNA-Werte im Hirngewebe nicht signifikant veränderte , in isolierten Pulmonalarterien (PA) jedoch keine nachweisbare Expression stattfand. Figur 3
gezeigte Linienprofil der relativen Fluoreszenzeinheiten über die Arterienwand vom Lumen bis zur Adventitia. Zusammenfassende Analyse des ASIC1-Ausdrucks in entweder (C) EC oder (D) SM von Lungenarterien aus Lungenschnitten. n = 5 Tiere pro Gruppe; analysiert mit einfaktorieller ANOVA und einzelne Gruppen verglichen mit Šídáks multiplen Vergleichstests. (E) PCR der Schwanz-DNA, die Cre-vermittelte LoxP-Rekombination und Exzision von gezieltem Asic1a in derselben MHC CreER - Asic1a fl/fl -Maus vor (-) und nach (+) Tamoxifen (TAM)-Behandlung zeigt. (F) Asic1a- mRNA-Expression in Hirngewebe und intrapulmonalen Arterien (PA) in MHC CreER - Asic1a fl/fl- Mäusen mit Vehikel (-) oder TAM (+). Gescannte Bilder der Blots wurden invertiert und hinsichtlich Helligkeit/Kontrast angepasst. Um die Rolle von PAEC und PASMC ASIC1a bei CH-induzierter pulmonaler Hypertonie zu bestimmen, haben wir drei verschiedene CH-Behandlungsparadigmen entwickelt, die in
Abbildung 4A
dargestellt sind : 1) Vehikel: Asic1a +/+ , Asic1a −/- , Tek Cre – Asic1a fl/fl und MHC CreER – Asic1a fl/fl Mäuse wurden mit Vehikel (Maisöl) behandelt und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 2) vorbeugend (pTAM): Asic1a fl/fl (pTAM) und MHC CreER – Asic1a fl/fl (pTAM) Mäuse wurden mit TAM behandelt (5 Tage) und 2 Wochen später 6 Wochen lang einer Kontrollgruppe oder CH ausgesetzt; 3) therapeutisch (tTAM): MHC CreER – Asic1a fl/fl (tTAM) Mäuse wurden 3 Wochen lang einer Kontrollgruppe oder CH ausgesetzt, um pulmonale Hypertonie festzustellen. Nach 3 Wochen CH wurden die Mäuse mit TAM behandelt (5 Tage mit con/CH-Exposition) und dann weitere 2 Wochen mit con/CH behandelt.
Tabelle 3
zeigt, dass die selektive Deletion von SM- oder EC- Asic1a den mittleren arteriellen Blutdruck oder die Herzfrequenz bei wachen Mäusen nicht signifikant veränderte und dem ähnelt, was wir zuvor bei Wildtyp-Mäusen beobachtet haben (
Detweiler et al., 2019
). Figur 4
Rechtsventrikulärer systolischer Druck (RVSP, mmHg) und (C) Fulton-Index (RV/LV + S) bei Asic1a +/+ , Asic1a −/- , Asic1a fl/fl (pTAM) , TekCre- Asic1a fl/fl , MHC CreER - Asic1a fl/fl , MHC CreER - Asic1a fl/fl (pTAM) oder MHC CreER - Asic1a fl/fl (tTAM) Mäusen unter Kontrollbedingungen (weiße Balken, Kreise) oder nach 6 Wochen CH (graue Balken, Quadrate). SM- Asic1a -Knockout wurde durch eine Behandlung mit Tamoxifen als vorbeugender (pTAM; vor Exposition gegenüber CH) oder therapeutischer Ansatz (tTAM; nach Auftreten von CH-induzierter pulmonaler Hypertonie) herbeigeführt. n = 5-8/Gruppe; analysiert durch zweiseitige ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p < 0,0001 für RVSP und RV/LV + S) wurden mit Šídáks Mehrfachvergleichstests untersucht; * p < 0,05 vs. Kontrolle; # p < 0,05 vs. jeweilige genetische Kontrolle. Tisch 3
Prozentuale Muskulaturisierung berechnet als Prozent SMA-Grenzwertfläche geteilt durch Gesamtarterienfläche basierend auf Arteriendurchmesser: <25 μm (n = ∼30 Gefäße von vier Tieren/Gruppe), 25–50 μm (n = ∼100 Gefäße von vier Tieren/Gruppe) oder 50–100 μm (n = ∼50 Gefäße von vier Tieren/Gruppe); analysiert mittels zweiseitiger ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p < 0,0001 für alle Gefäßdurchmesserbereiche) verglichen mit Šídáks Mehrfachvergleichstests. * p < 0,05 vs. Kontrolle; # p < 0,05 vs. (-) TAM; und τ p < 0,05 pTAM vs. tTAM. Die Proliferation von PAECs und PASMCs in MHC CreER - Asic1a fl/fl -Mäusen wurde zusätzlich mittels Immunfluoreszenz ausgewertet, um Ki-67-positive Zellen zu identifizieren (
Abbildung 6
). Die Deletion von SM- Asic1a hatte keinen signifikanten Einfluss auf den Prozentsatz positiver Ki-67-Kerne in PAECs, reduzierte jedoch den Prozentsatz proliferierender PASMCs signifikant (
Abbildung 6B
). Zusammen zeigen diese Daten, dass SM- Asic1a zur CH-induzierten Gefäßumgestaltung beiträgt, indem es sowohl zur Muskulaturisierung als auch zur PASMC-Proliferation beiträgt. Abbildung 6
Zusammenfassungsdaten für den Prozentsatz Ki-67-positiver Zellen in Endothelzellen (EC, blau) und glatten Muskelzellen (SMC, orange). Zusammenfassungsdaten für Kontrollmäuse werden nicht angezeigt, da keine Ki-67-positiven Zellen nachweisbar waren. n = 75 Gefäße von fünf Tieren (je 15 Gefäße) pro Gruppe; analysiert mit ungepaartem t -Test; # p < 0,05 vs. (-) TAM 3 Tage CH; ns = nicht signifikant. ASIC1 trägt zur Migration und Verbreitung von PASMC beiPASMCs wurden in vitro einer Hypoxie ausgesetzt , anschließend wurden Migration und Proliferation mittels Transwell-Assays bzw. Durchflusszytometrie für BrdU-positive Zellen untersucht. Hypoxie erhöhte den Prozentsatz migrierender mPASMCs von Asic1a +/+ -Mäusen signifikant , nicht jedoch von Asic1a −/- -Mäusen (
Abbildungen 7A, B ). Die Proliferation, gemessen anhand des BrdU-Einbaus, war bei mPASMCs von
Asic1a −/- -Mäusen unter Normoxie und nach Exposition gegenüber 24 und 48 h Hypoxie signifikant geringer (
Abbildung 7C
). Abbildung 7
zusammenfassende Daten für % migrierter mPASMC von Asic1a +/+ und Asic1a −/- Mäusen, die Normoxie (5 % CO2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO2 , 2 % O2 ; blaue Quadrate) für 24 hn = 6 Tiere/Gruppe ausgesetzt waren; * p < 0,05 vs. Hypoxie und # p < 0,05 Asic1a +/+ ; analysiert durch zweiseitige ANOVA. Signifikante Interaktionen zwischen den einzelnen Gruppen ( p = 0,0003) verglichen unter Verwendung von Šídáks Mehrfachvergleichstests. (C) Durchflusszytometrieanalyse des Prozentsatzes an mPASMC von Asic1a +/+ und Asic1a −/- Mäusen mit BrdU-Einbau unter Normoxie (0 h; 5 % CO 2 , 95 % Luft) oder Hypoxie (5 % CO 2 , 2 % O 2 ) für 24, 48 oder 72 h. Normoxische mPASMC von Asic1a +/+ wurden 72 h lang mit PDGF-BB (20 ng/ml) als positive Kontrolle inkubiert. n = fünf bis acht Tiere/Gruppe, zu jedem Zeitpunkt durch ungepaarte t-Tests analysiert, * p < 0,05. Um festzustellen, ob diese Erkenntnisse auf menschliche PASMCs (hPASMCs) übertragbar sind, haben wir die Auswirkungen von Hypoxie auf ASIC1-Expression, -Migration und -Proliferation in hPASMCs in Anwesenheit oder Abwesenheit einer ASIC1-Hemmung untersucht. Obwohl 12-stündige Hypoxie die Gesamtexpression von ASIC1 nicht veränderte (
Abbildung 8A, B
), vergrößerte sie die Plasmamembran (Zelloberfläche) im Vergleich zur zytosolischen Expression signifikant (
Abbildung 8C
). Dies steht im Einklang mit früheren Studien an 4 Wochen CH-exponierten Ratten, bei denen die Gesamtexpression von ASIC1 unverändert blieb, Hypoxie jedoch eine subzelluläre Translokation von ASIC1 zur Plasmamembran verursachte (
Herbert et al., 2016
,
2018
). Der Prozentsatz der Reinvasion von hPASMCs war nach 12-stündiger Hypoxie höher als nach Normoxie und wurde durch eine Vorbehandlung mit Amilorid oder PcTX1 verhindert (
Abbildung 8D, E
). Die durch Hypoxie induzierte Proliferation wurde in hPASMCs zusätzlich durch Amilorid und PcTX1 blockiert (
Abbildung 8F
). Zusammengenommen deuten diese Daten auf eine Beteiligung von ASIC1 an der durch Hypoxie vermittelten Proliferation und Migration von hPASMCs hin. Abbildung 8
Zusammenfassung der Daten, die die ASIC1-Expression (normalisiert auf die gesamte Spur des Coomassie-gefärbten Blots) in menschlichen PASMC (hPASMC) zeigen, die Normoxie (Norm, 5 % CO2 , 95 % Luft; weiße Balken/Kreise) oder 12 Stunden Hypoxie (Hyp, 5 % CO2 , 2 % O2 ; graue Balken/Quadrate) ausgesetzt waren. Die gescannten Bilder des Films wurden in Graustufen umgewandelt und Helligkeit/Kontrast angepasst. (C) Zusammenfassung der Daten, die die Western-Blot-Analyse der Expression von biotinyliertem (Plasmamembran-) und zytosolischem ASIC1-Protein in hPASMC zeigen, die Normoxie oder Hypoxie ausgesetzt waren. (D) Hellfeldbilder von hPASMC unmittelbar nach dem Kratzer (Basiszeit 0; obere Bildreihe) oder 12 Stunden nach der Exposition (untere Bildreihe) gegenüber Normoxie (5 % CO 2 , 95 % Luft; orange Kreise) oder Hypoxie (5 % CO 2 , 2 % O 2 ; blaue Quadrate) und (E) zusammenfassende Daten, die die prozentuale Reinvasion von hPASMC in den verletzten Bereich in Gegenwart von Vehikel, Amilorid oder PcTX1 zeigen. n = 8–11/Gruppe; analysiert mittels zweiseitiger ANOVA. Signifikante Wechselwirkungen zwischen den einzelnen Gruppen ( p = 0,0032) wurden mithilfe von Šídáks Mehrfachvergleichstests verglichen. (F) Zunahme der Zellzahl OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.