
- Beiträge: 1757
Sidebar
PH und Therapien, Management bei Corona
10 Mär 2023 12:00 #1733
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
PH und Therapien, Management bei Corona wurde erstellt von danny
www.dovepress.com/the-evolving-managemen...ulltext-article-VHRM
Die sich entwickelnden Management- und Behandlungsoptionen für Patienten mit pulmonaler Hypertonie: Aktuelle Erkenntnisse und Herausforderungen Zusammenfassung:Pulmonale Hypertonie kann sich als pulmonalarterienspezifischer Krankheitsprozess ohne erkennbare Ursache entwickeln oder im Zusammenhang mit anderen kardiopulmonalen und systemischen Erkrankungen auftreten. Die Weltgesundheitsorganisation (WHO) klassifiziert pulmonale hypertensive Erkrankungen auf der Grundlage der primären Mechanismen, die einen erhöhten pulmonalen Gefäßwiderstand verursachen. Eine wirksame Behandlung der pulmonalen Hypertonie beginnt mit der genauen Diagnose und Klassifizierung der Krankheit, um die geeignete Behandlung zu bestimmen. Die pulmonale arterielle Hypertonie (PAH) ist eine besonders herausfordernde Form der pulmonalen Hypertonie, da es sich um einen fortschreitenden, hyperproliferativen arteriellen Prozess handelt, der unbehandelt zu Rechtsherzversagen und Tod führt. In den letzten zwei Jahrzehnten Unser Verständnis der Pathobiologie und Genetik hinter PAH hat sich weiterentwickelt und zur Entwicklung mehrerer zielgerichteter Krankheitsmodifikatoren geführt, die die Hämodynamik und Lebensqualität verbessern. Effektive Risikomanagementstrategien und aggressivere Behandlungsprotokolle haben auch bessere Ergebnisse für Patienten mit PAH ermöglicht. Für Patienten, bei denen es unter medikamentöser Therapie zu einer fortschreitenden PAH kommt, bleibt die Lungentransplantation eine lebensrettende Option. Neuere Arbeiten waren auf die Entwicklung wirksamer Behandlungsstrategien für andere Formen von pulmonaler Hypertonie gerichtet, wie etwa chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) und pulmonaler Hypertonie aufgrund anderer Lungen- oder Herzerkrankungen. Die Entdeckung neuer Krankheitswege und Modifikatoren, die den Lungenkreislauf beeinflussen, ist ein fortlaufendes Gebiet intensiver Forschung.
Schlüsselwörter: pulmonale arterielle Hypertonie, chronisch thromboembolische pulmonale Hypertonie, Stickoxidweg, Endothelin-Antagonismus, Prostacyclin-AnalogonEinführungPulmonale Hypertonie (PH) ist definiert als ein mittlerer Pulmonalarteriendruck (PAP) größer oder gleich 20 mmHg, gemessen durch Rechtsherzkatheter. 1 , 3 Mehrere Mechanismen hinter der Entwicklung eines erhöhten Lungenarteriendrucks haben dazu geführt, dass die Weltgesundheitsorganisation (WHO) pulmonale hypertensive Erkrankungen in fünf verschiedene Gruppen eingeteilt hat. 1 Eine der größten Herausforderungen bei der Annäherung an einen Patienten mit pulmonaler Hypertonie ist die Aufgabe, die Krankheit richtig zu diagnostizieren und zu klassifizieren, um die geeignete Behandlung zu bestimmen. In einigen Fällen ist die optimale Behandlung einer assoziierten Erkrankung die primäre Behandlung der Wahl, aber in anderen Situationen können gezielte Medikamente oder chirurgische Maßnahmen indiziert sein. In den letzten zwei Jahrzehnten wurden erhebliche wissenschaftliche Anstrengungen unternommen, um die Pathobiologie hinter der pulmonalen arteriellen Hypertonie (PAH) der WHO-Gruppe 1 zu verstehen. PAH ist eine Erkrankung, die die Lungenarterien betrifft und durch spezifische Veränderungen in der Arterienmorphologie und einen steigenden Lungengefäßwiderstand gekennzeichnet ist, die zu Rechtsherzversagen und Tod führen können. Pathophysiologische Wege, die an PAH beteiligt sind, wurden definiert. 2 , 3 Eine Reihe neuer Mechanismen und Mediatoren werden derzeit untersucht. Die Entdeckung spezifischer krankheitsverursachender Mechanismen hat die Entwicklung zielgerichteter Therapien gefördert, die bei der Kontrolle der fortschreitenden Arteriopathie und der Symptome von PAH wirksam sind. Mehrere Untersuchungen sind im Gange, um zu klären, ob dieselben zielgerichteten Modifikatoren bei der Behandlung von pulmonaler Hypertonie in anderen WHO-Gruppen von Vorteil sein könnten. Diese Übersichtsarbeit beschreibt die grundlegenden Mechanismen, die an der Klassifizierung von pulmonal-hypertensiven Erkrankungen beteiligt sind, geht detaillierter auf das aktuelle Verständnis der PAH-Pathobiologie ein, skizziert aktuelle Managementstrategien, um optimale Ergebnisse für Patienten mit PAH zu erzielen, und fasst mehr zusammen jüngsten Bemühungen, Lösungen für die Behandlung von PH in anderen WHO-Gruppen zu finden.
Diagnose und Klassifizierung von pulmonalen hypertensiven ErkrankungenDie pulmonale Hypertonie wurde erstmals 1973 während des 1. Weltsymposiums über pulmonale Hypertonie in Genf, Schweiz, von der Weltgesundheitsorganisation nach pathologischen und klinischen Merkmalen klassifiziert. 4 Das Klassifizierungssystem der WHO wurde im Laufe der Zeit weiter verfeinert, wobei die jüngsten Aktualisierungen vom 6. Weltsymposium im Jahr 2018 und den Leitlinien der ESC/ERS (European Society of Cardiology/European Respiratory Society) von 2022 für die Diagnose und Behandlung von pulmonaler Hypertonie stammen. 1 , 3 Die Klassifizierung basiert auf den primären Mechanismen, die einen erhöhten pulmonalarteriellen Druck verursachen ( Abbildung 1 ). Patienten mit PAH der WHO-Gruppe 1 haben eine ausgeprägte Arteriopathie, die durch übermäßige Proliferation der zellulären Komponenten der Gefäßwand, Hypertrophie der glatten Muskulatur, In-situ-Thrombose und Bildung plexiformer Läsionen gekennzeichnet ist, die das Gefäßlumen verschließen. 5 , 6 Hyperproliferative Veränderungen, die in dieser Gruppe auftreten, können sich auch in pulmonalvenösen Strukturen entwickeln. PH der WHO-Gruppe 2 resultiert in erster Linie aus steigenden postkapillären Drücken, wie sie bei Erkrankungen beobachtet werden, die die linke Herzseite betreffen, wie Kardiomyopathien oder Herzklappenerkrankungen. Ein dritter Mechanismus, der pulmonale Hypertonie verursacht, wird durch Hypoxie und damit verbundene Vasokonstriktion vermittelt. Hypoxische Vasokonstriktion in der WHO-Gruppe 3 PH kann mit großer Höhe, Schlafapnoe und anderen Lungenerkrankungen wie Lungenfibrose oder Emphysem einhergehen. Eine Kreislaufobstruktion, die aus thromboembolischen oder anderen embolischen Ereignissen resultiert, kann zu einer pulmonalen Hypertonie der WHO-Gruppe 4 führen. Die endgültige Klassifikation, WHO-Gruppe 5, besteht aus Erkrankungen, die mit pulmonaler Hypertonie assoziiert sind, ohne einheitliche mechanistische Merkmale.
 Abbildung 1 Klassifikation pulmonaler hypertensiver Erkrankungen.Abkürzungen : HF, Herzinsuffizienz; PAH, pulmonale arterielle Hypertonie; PCH, pulmonale kapillare Hämangiomatose; PH, pulmonale Hypertonie; PVOD, pulmonale Venenverschlusskrankheit. Hinweise : a Patienten mit vererbbarer PAH oder PAH im Zusammenhang mit Arzneimitteln und Toxinen können akute Ansprecher sein. b linksventrikuläre Ejektionsfraktion bei Herzinsuffizienz mit reduzierter Ejektionsfraktion: < 40 %; bei Herzinsuffizienz mit leicht reduzierter Ejektionsfraktion: 41–49 %. c Andere Ursachen für Pulmonalarterienobstruktionen können sein: Sarkom (hoch- oder mittelgradiges oder Angiosarkom), andere bösartige Tumore (z. B. Nierenkarzinom, Uteruskarzinom, Keimzelltumoren der Hoden), nicht bösartige Tumore (z. B. Uterusleiomyom). ), Arteriitis ohne Bindegewebserkrankung, angeborene Pulmonalarterienstenosen und Hydatidose. d Einschließlich angeborener und erworbener chronischer hämolytischer Anämie und chronischer myeloproliferativer Erkrankungen. eEinschließlich Sarkoidose, pulmonale Langerhan-Zell-Histiozytose und Neurofibromatose Typ 1. f Einschließlich Glykogenspeicherkrankheiten und Morbus Gaucher. Wiedergabe mit freundlicher Genehmigung von Humbert M., Kovacs G., Hoeper MM, et al. 2022 ESC/ERS Leitlinien für die Diagnose und Behandlung von pulmonaler Hypertonie. Eur Respir J . 6. Jan. 2023;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society. 3 Das Vorliegen einer pulmonalen Hypertonie kann aufgrund der Anamnese, der Symptome und der Befunde im Elektrokardiogramm oder Röntgenaufnahmen des Brustkorbs vermutet werden.
7
Eine sorgfältige Anamnese anderer Krankheitszustände ist für die Bestimmung kausaler Zusammenhänge und die Einstufung nach WHO-Kriterien unerlässlich. Bindegewebserkrankungen, portale Hypertonie, Infektion mit dem humanen Immundefizienzvirus, angeborene Herzerkrankungen und bestimmte Expositionen gegenüber Arzneimitteln oder Toxinen wurden mit der Entwicklung der gleichen Arteriopathie in Verbindung gebracht, die bei PAH beobachtet wird.
8
Die Symptome sind unspezifisch und überschneiden sich mit denen vieler anderer kardiopulmonaler Erkrankungen. Daten aus dem Register REVEAL (Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Management) zeigen, dass Kurzatmigkeit und Müdigkeit die häufigsten Symptome sind. Schmerzen in der Brust werden oft festgestellt, sind aber weniger verbreitet. Ödeme und Synkopen sind ominöse Symptome, die normalerweise auf ein fortgeschritteneres Stadium der pulmonalen Hypertonie hinweisen.
9
,
10
Zu den körperlichen Manifestationen der pulmonalen Hypertonie bei der Untersuchung können ein verstärkter zweiter pulmonaler Herzton, ein systolisches Geräusch aufgrund einer Trikuspidalinsuffizienz und ein rechtsventrikulärer Lift gehören. Jugularvenöse Distension, Aszites und Ödeme sind Manifestationen einer fortgeschrittenen pulmonalen Gefäßerkrankung.
11
Anzeichen einer rechtsventrikulären Hypertrophie oder Rechtsachsenabweichung im EKG sind Indikatoren für eine zugrunde liegende pulmonale Hypertonie, ebenso wie vergrößerte zentrale Lungenarterien, periphere Gefäßbeschneidung und Obliteration des retrosternalen freien Raums auf Röntgenaufnahmen des Brustkorbs.
7
,
12
Wenn Anamnese, Untersuchungsbefunde und grundlegende diagnostische Studien auf eine pulmonale Hypertonie hindeuten, kann der Verdacht durch ein weiteres Screening mit einem Echokardiogramm bestätigt werden.
13–15
Das standardmäßige transthorakale Echokardiogramm kann anhand der Geschwindigkeit der Trikuspidalinsuffizienz eine Schätzung des Pulmonalarteriendrucks liefern. Das Echokardiogramm ist auch hilfreich bei der Unterscheidung der Linksherzursachen für pulmonale Hypertonie und liefert nützliche prognostische Informationen aus der Beurteilung der rechtsventrikulären Eigenschaften und des Vorhandenseins oder Fehlens eines Perikardergusses. Nach der Echokardiographie beinhaltet der diagnostische Algorithmus einen Prozess zur Beseitigung der Ursachen der sekundären pulmonalen Hypertonie (
Abbildung 2
). Aktuelle Richtlinien empfehlen einen algorithmischen Ansatz, der Blutuntersuchungen zum Screening auf autoimmune Bindegewebserkrankungen, HIV-Infektionen, Leber- und Schilddrüsenfunktionsstörungen, Screening auf Schlafapnoe, CT-Scans zum Ausschluss einer fibrotischen oder emphysematösen parenchymalen Lungenerkrankung, einen VQ-Scan zur Eliminierung von thromboembolischen Erkrankungen und Lungenerkrankungen umfasst Funktionstests zum Nachweis obstruktiver oder restriktiver Lungenerkrankungen.
3
,
16
Abbildung 1 Klassifikation pulmonaler hypertensiver Erkrankungen.Abkürzungen : HF, Herzinsuffizienz; PAH, pulmonale arterielle Hypertonie; PCH, pulmonale kapillare Hämangiomatose; PH, pulmonale Hypertonie; PVOD, pulmonale Venenverschlusskrankheit. Hinweise : a Patienten mit vererbbarer PAH oder PAH im Zusammenhang mit Arzneimitteln und Toxinen können akute Ansprecher sein. b linksventrikuläre Ejektionsfraktion bei Herzinsuffizienz mit reduzierter Ejektionsfraktion: < 40 %; bei Herzinsuffizienz mit leicht reduzierter Ejektionsfraktion: 41–49 %. c Andere Ursachen für Pulmonalarterienobstruktionen können sein: Sarkom (hoch- oder mittelgradiges oder Angiosarkom), andere bösartige Tumore (z. B. Nierenkarzinom, Uteruskarzinom, Keimzelltumoren der Hoden), nicht bösartige Tumore (z. B. Uterusleiomyom). ), Arteriitis ohne Bindegewebserkrankung, angeborene Pulmonalarterienstenosen und Hydatidose. d Einschließlich angeborener und erworbener chronischer hämolytischer Anämie und chronischer myeloproliferativer Erkrankungen. eEinschließlich Sarkoidose, pulmonale Langerhan-Zell-Histiozytose und Neurofibromatose Typ 1. f Einschließlich Glykogenspeicherkrankheiten und Morbus Gaucher. Wiedergabe mit freundlicher Genehmigung von Humbert M., Kovacs G., Hoeper MM, et al. 2022 ESC/ERS Leitlinien für die Diagnose und Behandlung von pulmonaler Hypertonie. Eur Respir J . 6. Jan. 2023;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society. 3 Das Vorliegen einer pulmonalen Hypertonie kann aufgrund der Anamnese, der Symptome und der Befunde im Elektrokardiogramm oder Röntgenaufnahmen des Brustkorbs vermutet werden.
7
Eine sorgfältige Anamnese anderer Krankheitszustände ist für die Bestimmung kausaler Zusammenhänge und die Einstufung nach WHO-Kriterien unerlässlich. Bindegewebserkrankungen, portale Hypertonie, Infektion mit dem humanen Immundefizienzvirus, angeborene Herzerkrankungen und bestimmte Expositionen gegenüber Arzneimitteln oder Toxinen wurden mit der Entwicklung der gleichen Arteriopathie in Verbindung gebracht, die bei PAH beobachtet wird.
8
Die Symptome sind unspezifisch und überschneiden sich mit denen vieler anderer kardiopulmonaler Erkrankungen. Daten aus dem Register REVEAL (Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Management) zeigen, dass Kurzatmigkeit und Müdigkeit die häufigsten Symptome sind. Schmerzen in der Brust werden oft festgestellt, sind aber weniger verbreitet. Ödeme und Synkopen sind ominöse Symptome, die normalerweise auf ein fortgeschritteneres Stadium der pulmonalen Hypertonie hinweisen.
9
,
10
Zu den körperlichen Manifestationen der pulmonalen Hypertonie bei der Untersuchung können ein verstärkter zweiter pulmonaler Herzton, ein systolisches Geräusch aufgrund einer Trikuspidalinsuffizienz und ein rechtsventrikulärer Lift gehören. Jugularvenöse Distension, Aszites und Ödeme sind Manifestationen einer fortgeschrittenen pulmonalen Gefäßerkrankung.
11
Anzeichen einer rechtsventrikulären Hypertrophie oder Rechtsachsenabweichung im EKG sind Indikatoren für eine zugrunde liegende pulmonale Hypertonie, ebenso wie vergrößerte zentrale Lungenarterien, periphere Gefäßbeschneidung und Obliteration des retrosternalen freien Raums auf Röntgenaufnahmen des Brustkorbs.
7
,
12
Wenn Anamnese, Untersuchungsbefunde und grundlegende diagnostische Studien auf eine pulmonale Hypertonie hindeuten, kann der Verdacht durch ein weiteres Screening mit einem Echokardiogramm bestätigt werden.
13–15
Das standardmäßige transthorakale Echokardiogramm kann anhand der Geschwindigkeit der Trikuspidalinsuffizienz eine Schätzung des Pulmonalarteriendrucks liefern. Das Echokardiogramm ist auch hilfreich bei der Unterscheidung der Linksherzursachen für pulmonale Hypertonie und liefert nützliche prognostische Informationen aus der Beurteilung der rechtsventrikulären Eigenschaften und des Vorhandenseins oder Fehlens eines Perikardergusses. Nach der Echokardiographie beinhaltet der diagnostische Algorithmus einen Prozess zur Beseitigung der Ursachen der sekundären pulmonalen Hypertonie (
Abbildung 2
). Aktuelle Richtlinien empfehlen einen algorithmischen Ansatz, der Blutuntersuchungen zum Screening auf autoimmune Bindegewebserkrankungen, HIV-Infektionen, Leber- und Schilddrüsenfunktionsstörungen, Screening auf Schlafapnoe, CT-Scans zum Ausschluss einer fibrotischen oder emphysematösen parenchymalen Lungenerkrankung, einen VQ-Scan zur Eliminierung von thromboembolischen Erkrankungen und Lungenerkrankungen umfasst Funktionstests zum Nachweis obstruktiver oder restriktiver Lungenerkrankungen.
3
,
16
 Abbildung 2 Algorithmus zur Diagnose der pulmonalen arteriellen Hypertonie.Abkürzungen : EKG, Elektrokardiogramm; CXR, Röntgen-Thorax; VTR, Geschwindigkeits-Trikuspidalinsuffizienz; LV, linker Ventrikel; PH, pulmonale Hypertonie; PAH, pulmonale arterielle Hypertonie; VQ, Ventilationsperfusion; CT, CT-Scan; RHC, Rechtsherzkatheterisierung; mPAP, mittlerer Lungenarteriendruck; CWP, Kapillarkeildruck; PVR, pulmonaler Gefäßwiderstand; WU, Holzeinheiten; HIV, menschliches Immunschwächevirus. Anmerkungen : Swisher JW und Kailash S. Fortschritte bei der Behandlung von pulmonaler Hypertonie im Zusammenhang mit systemischer Sklerose. In: Neue Erkenntnisse zur systemischen Sklerose . (Michal Tomcik, Hrsg.) InTech Open, London, UK.© 2019 Der/die Autor(en). Lizenznehmer IntechOpen. Dieses Kapitel wird unter den Bedingungen der Creative Commons Attribution 3.0 License verteilt, die die uneingeschränkte Nutzung, Verbreitung und Vervielfältigung in jedem Medium erlaubt.
17
Die Rechtsherzkatheterisierung bietet die genaueste Messung des pulmonalarteriellen Drucks und ist die abschließende diagnostische Bestätigungsstudie. Eine vollständige Rechtsherzstudie bestimmt auch die Vasoreaktivität, bewertet den postkapillären Lungenkreislaufdruck, identifiziert intrakardiale Shunts von links nach rechts und liefert wertvolle prognostische Daten.
18
Die traditionelle hämodynamische Definition der präkapillären PAH wurde auf dem 6. World Symposium on Pulmonary Hypertension im Jahr 2018 aktualisiert, um Folgendes einzuschließen: a) mittlerer PA-Druck > 20 mmHg, b) pulmonaler kapillarer Wedge-Druck ≤ 15 mmHg und c) pulmonaler Gefäßwiderstand (PVR). ) ≥3 WU (Holzeinheiten).
1
Die kürzlich veröffentlichten ESC/ERS-Richtlinien für die Diagnose und Behandlung von pulmonaler Hypertonie von 2022 modifizieren diese hämodynamische Definition weiter, indem sie das PVR-Kriterium auf einen beliebigen Wert > 2 WU senken.
3
Abbildung 3
enthält eine Zusammenfassung der neuesten hämodynamischen Definitionen für PH aus den ESC/ERS-Richtlinien. Bei Patienten mit idiopathischer, erblicher oder arzneimittelbedingter PAH identifiziert ein Vasoreaktivitätstest eine kleine Untergruppe von PAH-Patienten, die möglicherweise gut auf eine Behandlung mit Kalziumblockern ansprechen. Vasoreaktivitätstests können mit inhaliertem Stickstoffmonoxid, Adenosin oder intravenösem Epoprostenol durchgeführt werden. Eine positive Vasoreaktivitätsstudie wird durch eine Abnahme des mittleren PAP um mindestens 10 mmHg und einen absoluten Wert des mittleren PAP von weniger als 40 mmHg bestimmt.
19
Begleitende Tests mit körperlicher Betätigung, Anheben des geraden Beins oder Flüssigkeitsprovokation während der Rechtsherzkatheterisierung können eine postkapilläre pulmonale Hypertonie demaskieren, die bei Herzinsuffizienz und erhaltener Ejektionsfraktion beobachtet wird.
20–22
Bestimmte Daten, die während der Rechtsherzkatheterisierung erhoben werden, insbesondere der rechtsatriale Druck und der Herzindex, haben sich als wichtige Prädiktoren für das Langzeitüberleben erwiesen.
23
,
24
Abbildung 2 Algorithmus zur Diagnose der pulmonalen arteriellen Hypertonie.Abkürzungen : EKG, Elektrokardiogramm; CXR, Röntgen-Thorax; VTR, Geschwindigkeits-Trikuspidalinsuffizienz; LV, linker Ventrikel; PH, pulmonale Hypertonie; PAH, pulmonale arterielle Hypertonie; VQ, Ventilationsperfusion; CT, CT-Scan; RHC, Rechtsherzkatheterisierung; mPAP, mittlerer Lungenarteriendruck; CWP, Kapillarkeildruck; PVR, pulmonaler Gefäßwiderstand; WU, Holzeinheiten; HIV, menschliches Immunschwächevirus. Anmerkungen : Swisher JW und Kailash S. Fortschritte bei der Behandlung von pulmonaler Hypertonie im Zusammenhang mit systemischer Sklerose. In: Neue Erkenntnisse zur systemischen Sklerose . (Michal Tomcik, Hrsg.) InTech Open, London, UK.© 2019 Der/die Autor(en). Lizenznehmer IntechOpen. Dieses Kapitel wird unter den Bedingungen der Creative Commons Attribution 3.0 License verteilt, die die uneingeschränkte Nutzung, Verbreitung und Vervielfältigung in jedem Medium erlaubt.
17
Die Rechtsherzkatheterisierung bietet die genaueste Messung des pulmonalarteriellen Drucks und ist die abschließende diagnostische Bestätigungsstudie. Eine vollständige Rechtsherzstudie bestimmt auch die Vasoreaktivität, bewertet den postkapillären Lungenkreislaufdruck, identifiziert intrakardiale Shunts von links nach rechts und liefert wertvolle prognostische Daten.
18
Die traditionelle hämodynamische Definition der präkapillären PAH wurde auf dem 6. World Symposium on Pulmonary Hypertension im Jahr 2018 aktualisiert, um Folgendes einzuschließen: a) mittlerer PA-Druck > 20 mmHg, b) pulmonaler kapillarer Wedge-Druck ≤ 15 mmHg und c) pulmonaler Gefäßwiderstand (PVR). ) ≥3 WU (Holzeinheiten).
1
Die kürzlich veröffentlichten ESC/ERS-Richtlinien für die Diagnose und Behandlung von pulmonaler Hypertonie von 2022 modifizieren diese hämodynamische Definition weiter, indem sie das PVR-Kriterium auf einen beliebigen Wert > 2 WU senken.
3
Abbildung 3
enthält eine Zusammenfassung der neuesten hämodynamischen Definitionen für PH aus den ESC/ERS-Richtlinien. Bei Patienten mit idiopathischer, erblicher oder arzneimittelbedingter PAH identifiziert ein Vasoreaktivitätstest eine kleine Untergruppe von PAH-Patienten, die möglicherweise gut auf eine Behandlung mit Kalziumblockern ansprechen. Vasoreaktivitätstests können mit inhaliertem Stickstoffmonoxid, Adenosin oder intravenösem Epoprostenol durchgeführt werden. Eine positive Vasoreaktivitätsstudie wird durch eine Abnahme des mittleren PAP um mindestens 10 mmHg und einen absoluten Wert des mittleren PAP von weniger als 40 mmHg bestimmt.
19
Begleitende Tests mit körperlicher Betätigung, Anheben des geraden Beins oder Flüssigkeitsprovokation während der Rechtsherzkatheterisierung können eine postkapilläre pulmonale Hypertonie demaskieren, die bei Herzinsuffizienz und erhaltener Ejektionsfraktion beobachtet wird.
20–22
Bestimmte Daten, die während der Rechtsherzkatheterisierung erhoben werden, insbesondere der rechtsatriale Druck und der Herzindex, haben sich als wichtige Prädiktoren für das Langzeitüberleben erwiesen.
23
,
24
 Abbildung 3 Hämodynamische Definitionen der pulmonalen Hypertonie.Abkürzungen : mPAP, mittlerer pulmonalarterieller Druck; PAWP, pulmonalarterieller Keildruck; PVR, pulmonaler Gefäßwiderstand; WU, Holzeinheiten. Anmerkungen : Reproduziert mit freundlicher Genehmigung von Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Leitlinien für die Diagnose und Behandlung von pulmonaler Hypertonie. Eur Respir J . 6. Jan. 2023;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society. 3 . EpidemiologieDie meisten demografischen Daten zur pulmonalen Hypertonie beziehen sich auf Patienten mit PAH der WHO-Gruppe 1. In anderen WHO-Gruppen sind nur wenige detaillierte Informationen zur Epidemiologie verfügbar. Der vielleicht umfassendste Bericht zur Epidemiologie in den WHO-Gruppen 1 bis 4 stammt aus einer großen bevölkerungsbezogenen Kohortenstudie in Ontario, Kanada.
25
Die Daten zu Inzidenz, Prävalenz, Komorbiditäten und Mortalität in diesem Bericht wurden von 1993 bis 2012 aus Aufzeichnungen über Krankenhausaufenthalte, Notaufnahmen und Krankenversicherungen für 50.529 Einwohner von Ontario extrahiert. Es ist wichtig, auf die Zuverlässigkeit der Daten in diesem Bericht hinzuweisen war abhängig von der Genauigkeit der diagnostischen Kodierung. Darüber hinaus wurden nur 40,9 % der Patienten mit Zwischenfällen in dieser Kohorte einer detaillierten diagnostischen Abklärung unterzogen, die eine Rechtsherzkatheterisierung umfasste. Unter Berücksichtigung dieser Einschränkungen ergab die Untersuchung, dass das Durchschnittsalter der Patienten mit pulmonaler Hypertonie jeglicher Ätiologie 68,5 Jahre betrug und 54,5 % weiblich waren. Die Gesamtgruppe umfasste 13,8 % der WHO-Gruppe 1, 68,5 % der Gruppe 2, 47 % der Gruppe 3 und 9 % der Gruppe 4 PH. Gemischte Merkmale der WHO-Gruppe wurden in 35 vorgeschlagen. 4 % der Patienten mit Diagnosecodes, die mehr als eine WHO-Gruppe bezeichnen. Diese Studie zeigte einen Anstieg sowohl der Inzidenz als auch der Prävalenz von PH während des Jahrzehnts von 2002 bis 2012. Die Sterblichkeitsraten für die gesamte PH-Population betrugen 12,8 %, 35,9 % und 61,5 % nach 30 Tagen, 1 Jahr bzw. 5 Jahren. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist.
26
Bevölkerungsregister aus Frankreich und den Vereinigten Staaten haben die epidemiologische Zusammensetzung der PAH der WHO-Gruppe 1 veranschaulicht.
27
,
28
Wie bereits erwähnt, kann die pulmonale arterielle Hypertonie abhängig davon, ob sie erblich oder idiopathisch ist oder mit anderen Erkrankungen assoziiert ist, die als Risikofaktoren für PAH gelten, in Unterklassen eingeteilt werden. Entitäten, die als Risikofaktoren für die Entwicklung von PAH angesehen werden, umfassen Arzneimittel- und Toxinexposition, Bindegewebserkrankungen, HIV-Infektion, portale Hypertonie, angeborene Herzerkrankungen und Schistosomiasis. Die Berichte über Inzidenz, Prävalenz, Mortalität und andere Merkmale von PAH weisen je nach geografischen Unterschieden in den Zielpopulationen, wirtschaftlichen Determinanten, Unterschieden in den Gesundheitssystemen und der klinischen Praxis große Unterschiede auf.
29–33
Die Quelle für die Datenerfassung kann ebenfalls ein wichtiger Faktor sein, der zur Variabilität beiträgt. Eine kürzlich durchgeführte umfassende Literaturrecherche zu PAH der WHO-Gruppe 1 und chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) der WHO-Gruppe 4 unterstreicht diesen Punkt.
29
Basierend auf Veröffentlichungen, die Daten aus nationalen Registern, klinischen Datenbanken und Anspruchs-/Verwaltungsdatenbanken widerspiegeln, hatten Patienten mit PAH ein Durchschnittsalter von 43 bis 76 Jahren, von denen 55 % bis 81 % weiblich waren. Die Inzidenz von PAH betrug 1,5 bis 32 Fälle pro Million der Allgemeinbevölkerung und die Prävalenz reichte von 12,4 bis 268 Fällen pro Million. In ähnlicher Weise wurde bei CTEPH eine große Variabilität mit einem Durchschnittsalter bei Diagnose von 58 bis 73 Jahren und 37 % bis 70 % Frauen berichtet. Die Inzidenz von CTEPH aus dieser Übersicht reichte von 0,9 bis 39 Fällen pro Million Einwohner und die Prävalenz reichte von 14,5 bis 144 pro Million.
Abbildung 3 Hämodynamische Definitionen der pulmonalen Hypertonie.Abkürzungen : mPAP, mittlerer pulmonalarterieller Druck; PAWP, pulmonalarterieller Keildruck; PVR, pulmonaler Gefäßwiderstand; WU, Holzeinheiten. Anmerkungen : Reproduziert mit freundlicher Genehmigung von Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Leitlinien für die Diagnose und Behandlung von pulmonaler Hypertonie. Eur Respir J . 6. Jan. 2023;61(1):2200879. Copyright 2023 European Society of Cardiology & European Respiratory Society. 3 . EpidemiologieDie meisten demografischen Daten zur pulmonalen Hypertonie beziehen sich auf Patienten mit PAH der WHO-Gruppe 1. In anderen WHO-Gruppen sind nur wenige detaillierte Informationen zur Epidemiologie verfügbar. Der vielleicht umfassendste Bericht zur Epidemiologie in den WHO-Gruppen 1 bis 4 stammt aus einer großen bevölkerungsbezogenen Kohortenstudie in Ontario, Kanada.
25
Die Daten zu Inzidenz, Prävalenz, Komorbiditäten und Mortalität in diesem Bericht wurden von 1993 bis 2012 aus Aufzeichnungen über Krankenhausaufenthalte, Notaufnahmen und Krankenversicherungen für 50.529 Einwohner von Ontario extrahiert. Es ist wichtig, auf die Zuverlässigkeit der Daten in diesem Bericht hinzuweisen war abhängig von der Genauigkeit der diagnostischen Kodierung. Darüber hinaus wurden nur 40,9 % der Patienten mit Zwischenfällen in dieser Kohorte einer detaillierten diagnostischen Abklärung unterzogen, die eine Rechtsherzkatheterisierung umfasste. Unter Berücksichtigung dieser Einschränkungen ergab die Untersuchung, dass das Durchschnittsalter der Patienten mit pulmonaler Hypertonie jeglicher Ätiologie 68,5 Jahre betrug und 54,5 % weiblich waren. Die Gesamtgruppe umfasste 13,8 % der WHO-Gruppe 1, 68,5 % der Gruppe 2, 47 % der Gruppe 3 und 9 % der Gruppe 4 PH. Gemischte Merkmale der WHO-Gruppe wurden in 35 vorgeschlagen. 4 % der Patienten mit Diagnosecodes, die mehr als eine WHO-Gruppe bezeichnen. Diese Studie zeigte einen Anstieg sowohl der Inzidenz als auch der Prävalenz von PH während des Jahrzehnts von 2002 bis 2012. Die Sterblichkeitsraten für die gesamte PH-Population betrugen 12,8 %, 35,9 % und 61,5 % nach 30 Tagen, 1 Jahr bzw. 5 Jahren. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist. Patienten mit PH der WHO-Gruppe 1 hatten die niedrigste Sterblichkeitsrate, während Patienten mit WHO-Gruppe 2 und 3 das höchste Sterberisiko hatten. Andere Berichte weisen auch darauf hin, dass Linksherzerkrankungen die häufigste Ursache für PH sind, während COPD weltweit die zweithäufigste ist.
26
Bevölkerungsregister aus Frankreich und den Vereinigten Staaten haben die epidemiologische Zusammensetzung der PAH der WHO-Gruppe 1 veranschaulicht.
27
,
28
Wie bereits erwähnt, kann die pulmonale arterielle Hypertonie abhängig davon, ob sie erblich oder idiopathisch ist oder mit anderen Erkrankungen assoziiert ist, die als Risikofaktoren für PAH gelten, in Unterklassen eingeteilt werden. Entitäten, die als Risikofaktoren für die Entwicklung von PAH angesehen werden, umfassen Arzneimittel- und Toxinexposition, Bindegewebserkrankungen, HIV-Infektion, portale Hypertonie, angeborene Herzerkrankungen und Schistosomiasis. Die Berichte über Inzidenz, Prävalenz, Mortalität und andere Merkmale von PAH weisen je nach geografischen Unterschieden in den Zielpopulationen, wirtschaftlichen Determinanten, Unterschieden in den Gesundheitssystemen und der klinischen Praxis große Unterschiede auf.
29–33
Die Quelle für die Datenerfassung kann ebenfalls ein wichtiger Faktor sein, der zur Variabilität beiträgt. Eine kürzlich durchgeführte umfassende Literaturrecherche zu PAH der WHO-Gruppe 1 und chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) der WHO-Gruppe 4 unterstreicht diesen Punkt.
29
Basierend auf Veröffentlichungen, die Daten aus nationalen Registern, klinischen Datenbanken und Anspruchs-/Verwaltungsdatenbanken widerspiegeln, hatten Patienten mit PAH ein Durchschnittsalter von 43 bis 76 Jahren, von denen 55 % bis 81 % weiblich waren. Die Inzidenz von PAH betrug 1,5 bis 32 Fälle pro Million der Allgemeinbevölkerung und die Prävalenz reichte von 12,4 bis 268 Fällen pro Million. In ähnlicher Weise wurde bei CTEPH eine große Variabilität mit einem Durchschnittsalter bei Diagnose von 58 bis 73 Jahren und 37 % bis 70 % Frauen berichtet. Die Inzidenz von CTEPH aus dieser Übersicht reichte von 0,9 bis 39 Fällen pro Million Einwohner und die Prävalenz reichte von 14,5 bis 144 pro Million.
Pathophysiologie der pulmonalen arteriellen Hypertonie (WHO-Gruppe 1)Patienten, die als PAH der WHO-Gruppe 1 klassifiziert sind, teilen eine Vaskulopathie der kleinen präkapillaren Arterien, die durch übermäßige Endothelproliferation, Proliferation und Hypertrophie der glatten Muskulatur, In-situ-Thrombose und Bildung von vaskulären plexiformen Läsionen gekennzeichnet ist. 5 , 34 Diese vaskulären Veränderungen können auch in den postkapillaren Gefäßen bei bestimmten Erkrankungen wie systemischer Sklerose und pulmonaler Venenverschlusskrankheit gefunden werden. Neben der charakteristischen Umgestaltung des Lungengefäßsystems kommt es zu einem Verlust präkapillarer Arterien und einer übertriebenen Infiltration des perivaskulären Raums mit Entzündungszellen. 2 Die Pathogenese dieser Veränderungen ist ein komplexer Prozess mit Hinweisen auf genetische Faktoren, Zytokine und Wachstumsfaktoren, Ionenkanaldysfunktion und Gefäßverletzung mit endothelialer Dysfunktion, die zu einem Ungleichgewicht der endogenen vasomotorischen Regulation und der Zellproliferation im pulmonalen Gefäßbett führt. 2 , 35 , 36 Eine genetische Grundlage für PAH wurde erstmals im Jahr 2000 mit der Entdeckung einer Genmutation des knochenmorphogenetischen Proteinrezeptors II (BMPR2) bei Patienten mit vererbbarer PAH aufgedeckt. 37 , 38 Diese Mutation wird bei 70–80 % der Patienten mit erblicher PAH und bei 15–25 % der Patienten mit idiopathischer PAH identifiziert. 39 , 40 BMPR2 ist ein Mitglied der Gen-Superfamilie des transformierenden Wachstumsfaktors Beta (TGF-") und dient dazu, die Proliferation glatter Muskelzellen und die Apoptose von Endothelzellen zu begrenzen.
41
Sequenzvariationen in Genen, die für BMPR2-verwandte nachgeschaltete SMAD (Suppressor of Mothers Against Decapentaplegic)-Signaltransduktoren (SMAD1, SMAD4, SMAD8, SMAD9) codieren, wurden ebenfalls mit der Pathogenese von PAH in Verbindung gebracht.
42–44
Mutationen, an denen Activin-Rezeptor-ähnliche Kinase 1 (ACVRL1 oder ALK1) und Endoglin (ENG) beteiligt sind, machen 80 % der Fälle von hereditärer hämorrhagischer Teleangiektasie (HHT) aus, wobei 15–20 % dieser Patienten auch eine pulmonale Hypertonie entwickeln.
45
,
46
Andere PAH-verbundene Mutationen, die nichts mit der BMPR2-Signalgebung zu tun haben, beinhalten KCNK3 und CAV1. KCNK3 (Kaliumkanal-Unterfamilie K-Mitglied 3) codiert einen pH-empfindlichen Kaliumkanal, der den Gefäßtonus beeinflusst.
47
CAV1 (Caveolin-1) kodiert für ein Membranprotein, das für die Bildung von Lipidflößen oder Caveoli wesentlich ist, die zur Positionierung von BMP-Rezeptoren dienen.
48
Bis zu 25 % der sporadischen Fälle von pulmonaler kapillärer Hämangiomatose und pulmonaler venöser Verschlusskrankheit wurden Mutationen in EIF2AK4 (eukaryotischer Translationsinitiationsfaktor 2 Alpha-Kinase 4) zugeschrieben.
49
,
50
Neben Mutationen des Genoms standen auch epigenetische Mechanismen, die die Zellfunktion beeinflussen, im Fokus der neueren Forschung. Diese Mechanismen können DNA-Methylierung, Mikro-RNAs oder Modifikation von Histonproteinen umfassen, die die Expression von Wachstumsfaktorspiegeln oder Genexpression verändern und dadurch Zellwachstum und -proliferation beeinflussen.
51–54
Die Infiltration von Entzündungszellen wird in der Nähe von Bereichen des Gefäßumbaus bei PAH beobachtet, was das Interesse an der potenziellen Rolle von Zytokinen und Wachstumsfaktoren im Prozess der Gefäßerkrankung weckt. Es ist unklar, ob das Vorhandensein von Entzündungszellinfiltraten eine Folge von Hypoxie darstellt oder ob Entzündungsmediatoren eine Gefäßzellschädigung und -funktionsstörung fördern. Die Untersuchung vaskulärer Läsionen hat eine Invasion von Lymphozyten, Makrophagen, Mastzellen und dendritischen Zellen gezeigt.
55–57
Während in den Lungen von Patienten mit idiopathischer PAH ein Mangel an regulatorischen T-Zellen festgestellt wurde,
58
deuten eine Ausdehnung des ektopischen pulmonalen Lymphgewebes und das Vorhandensein von Autoantikörpern auf eine übermäßige B-Zell-Aktivierung hin.
59
Ektopisches Gewebe in der Nähe von Lungenarterien bei PAH kann mit dem Gefäßumbauprozess in Verbindung gebracht werden. Dendritische Zellen setzen Zytokine frei, die T- und B-Lymphozyten anziehen, ihr Überleben verbessern und zu einer entzündlichen Umgebung beitragen.
60
Makrophagen, die sich in der Nähe von Gefäßläsionen befinden, sind anfällig für die Wirkungen von IL-6, das von aktivierten Adventitia-Fibroblasten freigesetzt wird, und nehmen einen Phänotyp mit proinflammatorischen Eigenschaften an.
61
Makrophagen sind eine Quelle des aus Blutplättchen stammenden Wachstumsfaktors (PDGF), der ein potentes Mitogen und Chemoattraktor für Gefäßzellen ist.
62
Obwohl die genaue Rolle der Immunzellbiologie und der Entzündung bei der Pathogenese von PAH noch definiert werden muss, spricht das Vorhandensein ähnlicher pulmonaler Gefäßveränderungen sowohl bei PAH als auch bei einer signifikanten Anzahl von Patienten mit primären Autoimmunerkrankungen für eine Rolle von immunzellvermittelten Ereignissen in Umbau der Lungengefäße.Abgesehen von der Auswirkung genetischer und immunologischer oder entzündlicher Faktoren auf die strukturelle Umgestaltung gibt es Hinweise darauf, dass eine Endothelverletzung und -dysfunktion Ungleichgewichte in der Produktion von endogenen Mediatoren des Gefäßtonus, der Blutplättchenaggregation und der Zellproliferation verursachen. Immunhistochemische Studien haben signifikante Reduktionen der Stickoxid-Synthase- und Prostacyclin-Synthase-Spiegel im Lungengefäßendothel gezeigt, wo diese Enzyme eine entscheidende Rolle bei der Produktion von Stickoxid und Prostacyclin spielen, die beide gefäßerweiternde und antiproliferative Wirkungen auf Lungengefäßzellen haben.
63
,
64
Während die Produktion von Stickoxid und Prostacyclin durch pulmonalvaskuläres Endothel reduziert ist, ist die Produktion von Endothelin-1 durch Endothelzellen bei PAH erhöht.
65
Endothelin-1 fördert gegensätzliche Eigenschaften, einschließlich Vasokonstriktion und Zellproliferation. Endothelin-1, Survivin und vaskulärer endothelialer Wachstumsfaktor (VEGF) wurden in vaskulären plexiformen Läsionen isoliert und es wird angenommen, dass sie die Proliferation von Endothelzellen und glatten Muskelzellen verstärken, während sie die Apoptose hemmen.
65–67
Die Thromboxanproduktion durch das Lungenendothel wird ebenfalls erhöht, was zu einer Vasokonstriktion und einer In-situ-Thrombusbildung in den Lungenarterien führt.
68
Andere Faktoren mit potenzieller Rolle bei der Pathogenese von PAH sind Serotonin, Autoantikörper, dysfunktionale spannungsgesteuerte Kaliumkanäle und krebsähnliche Muster der Zellproliferation und Apoptoseresistenz. Serotonin kann die Vasokonstriktion und den Gefäßumbau fördern, indem es die Proliferation glatter Muskelzellen (SMC) und Fibroblasten stimuliert.
69–71
Die Expression von Anti-Endothelzellen- und Anti-Fibroblasten-Antikörpern gegen spezifische Zielantigene wurde bei idiopathischer und Sklerodermie-assoziierter PAH beobachtet, obwohl die Rolle dieser Autoimmuneffektoren bei der Pathogenese von PAH unklar ist.
72
,
73
Die Proliferation glatter Muskelzellen kann durch Serotonin-Transporter-Aktivierung des Thrombozyten-Wachstumsfaktor-beta (PDGF--Rezeptors stimuliert werden.
74
Intrazelluläres Calcium der glatten Muskulatur reguliert nicht nur die Kontraktion, sondern auch die Proliferation und den Widerstand gegen Apoptose. Herunterregulierung und Dysfunktion von spannungsgesteuerten Kaliumkanälen beeinflussen die Membranpolarisation und erhöhen den Calciumeinstrom, was wiederum die Kontraktion glatter Muskelzellen fördert und die Proliferation verstärkt, indem Zellen in den Zellzyklus getrieben werden.
39
,
75
,
76
In Lungengefäßendothelzellen, SMCs und Fibroblasten wurde eine Reihe von krebsähnlichen zellulären Verhaltensweisen beobachtet, darunter die monoklonale Expansion von Endothelzellen bei idiopathischer PAH, das Vorhandensein instabiler kurzer DNA-Mikrosatellitensequenzen in plexiformen Läsionen und ein anhaltender hyperproliferativer und apoptoseresistenter Zustand wenn Endothelzellen aus der in vivo-Umgebung entfernt werden.
77–79
Endothelzellen der Lungengefäße, SMCs und Fibroblasten sind für die Energieerzeugung stärker von der Glykolyse abhängig.
80–82
Mitochondrien wechseln ähnlich wie Krebszellen von der Glukoseoxidation zur ungekoppelten aeroben Glykolyse und fördern so die Bildung von Vorläufern für die DNA-Synthese und die schnelle Zellproliferation.
83
Der vaskuläre Umbau bei PAH unterscheidet sich von Krebs durch die Tatsache, dass sich pulmonale Gefäßzellen nicht ohne Kontrolle klonal vermehren.Pathophysiologie hinter pulmonaler Hypertonie in anderen WHO-GruppenObwohl angenommen wird, dass pulmonale Hypertonie in Nicht-PAH-WHO-Gruppen typischerweise durch Mechanismen entsteht, die für andere Krankheitsprozesse und ihre Auswirkungen auf den Lungenkreislauf spezifisch sind, gibt es Hinweise auf Ähnlichkeiten in der Pathophysiologie zwischen WHO-Gruppen, die auf Endothelschäden oder Dysfunktionen wie die in PAH.
35
Pulmonale Hypertonie bei Linksherzerkrankung (PH-LHD) wird als Folge der retrograden Übertragung eines erhöhten linksatrialen Drucks auf den Lungenkreislauf angesehen. Erhöhter linksatrialer Druck kann eine Folge einer systolischen oder diastolischen linksventrikulären Dysfunktion oder einer Herzklappenerkrankung sein. Bei einigen Patienten der WHO-Gruppe 2 kann sich in den distalen Pulmonalarterien und -venolen ein vaskulärer Umbau mit PAH-ähnlichen Merkmalen entwickeln und auch nach Korrektur der zugrunde liegenden Ursache der linksatrialen Hypertonie bestehen bleiben.
84
Als Beispiel wurde gezeigt, dass Patienten mit Herzklappenerkrankungen nach Herzklappenreparatur und Entfernung der Quelle des erhöhten postkapillären Drucks Anzeichen einer anhaltenden pulmonalen Hypertonie aufweisen.
85
Es wird angenommen, dass eine endotheliale Dysfunktion, die aus einem verlängerten retrograden Anstieg des pulmonalen Gefäßdrucks resultiert, viele der beobachteten Veränderungen in der Gefäßstruktur und -funktion vermittelt.
89
Erhöhte Endothelinspiegel im Plasma werden bei Patienten mit PH-LHD berichtet, und die Stickoxidproduktion ist bei Herzinsuffizienz reduziert.
87
,
90
Es ist bekannt, dass die pulmonale Vaskulopathie bei PH-LHD die Aktivierung von Fibroblasten, die Proliferation der vaskulären Intima und Media, die perivaskuläre Infiltration mit Entzündungszellen und die erhöhte Zytokin- und Wachstumsfaktorexpression umfasst, die alle den bei PAH beobachteten Merkmalen ähneln.
86–88
Diese Befunde korrelieren mit Beobachtungen einer Gefäßumgestaltung, die der bei PAH beobachteten ähnelt.Pulmonale Hypertonie der WHO-Gruppe 3 aufgrund einer Lungenerkrankung und Hypoxie wird oft als Folge einer hypoxischen Vasokonstriktion und des Verlusts kleiner Gefäße und Kapillaren angesehen. Obwohl dies zutrifft, wurde chronische Hypoxie auch mit der Aktivität mehrerer Mediatoren in Verbindung gebracht, die die Gefäßstruktur und -funktion ähnlich der bei PAH festgestellten Gefäßumgestaltung beeinflussen.
91
,
92
Zusätzlicher Sauerstoff kann hilfreich sein, um die hypoxische Vasokonstriktion in frühen Stadien umzukehren, aber wenn die Krankheit fortschreitet und die Hypoxie chronischer wird, wird der Gefäßumbau zu einer bedeutenderen Ursache für den steigenden pulmonalen Gefäßwiderstand. Wie bei PAH spielt die Endothelbiologie eine Schlüsselrolle bei der Reaktion der Lungengefäße auf Hypoxie und wird durch TGF-B1 und Hypoxie-induzierbarer Faktor 1-alpha (HIF-1a) Signalwege beeinflusst.
81
,
91
,
93
,
94
In Versuchsmodellen induziert chronische Hypoxie die Expression des vaskulären endothelialen Wachstumsfaktors A (VEGFA) und seines Rezeptors VEGFR2 über HIF-1a.
91
,
95
,
96
Hypoxie induziert auch die Expression von TGF-B1 und erhöht dadurch die Produktion von PDGF-B. Es wurde gezeigt, dass PDGF-B die VEGFA-Expression und damit die Hypoxie-induzierte Endothelproliferation fördert.
91
,
97
Chronische Hypoxie ist ein Stimulus für die Freisetzung von Serotonin, was wiederum die Vasokonstriktion stimuliert und die Endothelproliferation und die Hypertrophie der glatten Muskulatur fördert.
92
,
98
Es wird berichtet, dass Hypoxie die Stickoxidproduktion der Endothelzellen verringert und die Freisetzung von Endothelin erhöht.
92
,
99
Als klinisches Korrelat wurde eine beeinträchtigte Endothel-abhängige SMC-Relaxation bei Patienten mit COPD unterschiedlichen Schweregrades beobachtet.
100
Es besteht ein beträchtliches Interesse an der Beziehung zwischen Tabakrauchen und vaskulärem Umbau und pulmonaler Hypertonie bei chronischen Lungenerkrankungen. Das Rauchen an sich scheint mehrere Auswirkungen auf die Lungengefäßbiologie zu haben, einschließlich erhöhter VEGF-Expression, reduzierter Stickoxid-Synthase-Spiegel, erhöhter Infiltration von Entzündungszellen, Endothelhyperplasie und gestörter Mitochondrienfunktion.
101–103
Obwohl insgesamt kaum verstanden, ist klar, dass die Ursachen der pulmonalen Hypertonie bei Patienten mit chronischer Lungenerkrankung und Hypoxie viel tiefer gehen als hypoxische Vasokonstriktion und Verlust kleiner Gefäße.Die pulmonale Hypertonie ist eine bekannte Komplikation der akuten pulmonalen Thromboembolie. In der akuten Phase einer Thromboembolie kann der pulmonalvaskuläre Widerstand durch eine signifikante Gerinnselbelastung erhöht sein, die das pulmonalvaskuläre Bett verstopft. Typischerweise wird durch Thrombolyse und/oder Antikoagulation das obstruktive Gerinnsel beseitigt und der normale Gefäßwiderstand wiederhergestellt. Doch selbst bei kontinuierlicher Antikoagulation können Lungenperfusionsstörungen in über 50 % der Fälle länger als 3 Monate nach einem akuten Ereignis bestehen bleiben.
104
Ein kleiner Prozentsatz dieser Patienten entwickelt später eine chronisch thromboembolische pulmonale Hypertonie der WHO-Gruppe 4 (CTEPH). Studien schätzen die jährliche Inzidenz von CTEPH nach einer akuten Lungenembolie auf 0,4 % bis 6,2 %.
105
Die wahre Inzidenz ist jedoch ungewiss, da aktuelle Schätzungen der CTEPH-Inzidenz nach akuter LE Patienten umfassen können, die bereits vor dem akuten Ereignis eine chronische Thromboembolie hatten. CTEPH kann mehrere Monate oder Jahre nach kontinuierlicher Antikoagulation und ohne symptomatische rezidivierende akute Ereignisse diagnostiziert werden. Die pulmonale Hypertonie in dieser Population ist teilweise eine Folge eines sich nicht auflösenden Thrombus. Im Gegensatz zu dem charakteristischen frischen Gerinnsel bei akuter Lungenembolie besteht das chronisch flussbegrenzende Material bei CTEPH aus einem gelben fibrotischen Material, das fest an der Gefäßwand haftet und aus Kollagen, Elastin, Entzündungszellen und Rekanalisationsgefäßen besteht.
105
Es gibt mehrere vorgeschlagene Theorien, die sich auf die Nichtauflösung von thrombotischem Material bei Patienten beziehen, die CTEPH entwickeln. Wenn die thromboembolische Belastung groß ist, kann das intrinsische lytische System aufgrund unzureichender lytischer Kapazität oder Fähigkeit, die gesamte Gerinnselmasse zu erreichen, möglicherweise nicht in der Lage sein, eine vollständige Auflösung zu erreichen. Eine andere Möglichkeit besteht darin, dass Patienten, die behandelt werden, möglicherweise nicht ausreichend oder nicht lange genug antikoaguliert werden. Zugrunde liegende Autoimmunerkrankungen, Nicht-O-Blutgruppe, Splenektomie in der Vorgeschichte, ventrikulo-atriale Shunts, Schilddrüsenersatz und eine Vorgeschichte von Malignität waren in einer großen Studie, in der Patienten mit CTEPH mit Patienten mit anderen Formen der pulmonalen Hypertonie verglichen wurden, alle mit einem höheren CTEPH-Risiko verbunden.
106
Bei CTEPH wurde über höhere Konzentrationen mehrerer Entzündungsmarker und Entzündungsmediatoren berichtet, was darauf hindeutet, dass die zugrunde liegende Entzündung irgendwie beteiligt ist.
107
,
108
Mutationen, die abnormales Fibrinogen und damit eine abnormale Fibrinstruktur und Resistenz gegenüber Plasmin-vermittelter Lyse verursachen, wurden identifiziert.
109–111
Anomalien der Thrombozytenfunktion und dysfunktionale Angiogenese und Rekanalisation des Thrombus wurden ebenfalls vorgeschlagen, um die Nichtauflösung des Thrombus bei CTEPH zu erklären.
112
,
113
Die Nichtauflösung des Gerinnsels allein erklärt nicht die Entwicklung einer pulmonalen Hypertonie bei CTEPH. Ein Umbau kleiner Gefäße, ähnlich dem bei idiopathischer PAH beschriebenen, wird auch bei CTEPH beobachtet und betrifft Gefäße mit ungehindertem Fluss sowie solche, die distal von flussbegrenzenden Thromben liegen. Weiterhin erstrecken sich die Charakteristika der arteriellen Vaskulopathie dahingehend, Venolen und kleine Venen einzubeziehen. Die Entwicklung einer Vaskulopathie in unverstopften Arterien wurde als Folge eines umgeleiteten Flusses mit erhöhtem Druck und Scherbeanspruchung angesehen, was zu einer Endothelverletzung führt.
114
Dieser Mechanismus würde jedoch nicht die Entwicklung einer Arteriopathie distal zu obstruktiven Thromben erklären. Es wurden Anastamosen zwischen den Bronchial- und Pulmonalarterien identifiziert, die einen Fluss distal zu pulmonalarteriellen Obstruktionen ermöglichen können.
115
Es wird die Theorie aufgestellt, dass das Aussetzen kleiner Arterien distal des Thrombus gegenüber systemischem Druck den Gefäßumbau in diesen Arterien fördern kann. Abgesehen von diesen mechanischen Überlegungen gibt es Hinweise darauf, dass bei CTEPH auch molekulare Faktoren existieren, die den Gefäßumbau begünstigen. Es ist bekannt, dass die Spiegel des endogenen Vasodilatators Stickstoffmonoxid sowohl bei Patienten mit PAH als auch mit CTEPH reduziert sind.
116
Darüber hinaus sind die Spiegel des Stickstoffmonoxid-Synthase-Inhibitors, asymmetrisches Dimethylarginin, erhöht und können die günstigen gefäßerweiternden und antiproliferativen Wirkungen von Stickstoffmonoxid auf vaskulärer Ebene einschränken.
117
Erhöhte Endothelinspiegel wurden, wie bei PAH erwähnt, auch bei CTEPH-Patienten beobachtet.
118
Obwohl Thrombose das primäre Ereignis bei CTEPH ist, gibt es immer mehr Hinweise darauf, dass die Entwicklung von pulmonaler Hypertonie mit Abweichungen in einem komplexen molekularen Signalsystem einhergeht, das in vielerlei Hinsicht dem bei PAH beobachteten ähnelt.
und dient dazu, die Proliferation glatter Muskelzellen und die Apoptose von Endothelzellen zu begrenzen.
41
Sequenzvariationen in Genen, die für BMPR2-verwandte nachgeschaltete SMAD (Suppressor of Mothers Against Decapentaplegic)-Signaltransduktoren (SMAD1, SMAD4, SMAD8, SMAD9) codieren, wurden ebenfalls mit der Pathogenese von PAH in Verbindung gebracht.
42–44
Mutationen, an denen Activin-Rezeptor-ähnliche Kinase 1 (ACVRL1 oder ALK1) und Endoglin (ENG) beteiligt sind, machen 80 % der Fälle von hereditärer hämorrhagischer Teleangiektasie (HHT) aus, wobei 15–20 % dieser Patienten auch eine pulmonale Hypertonie entwickeln.
45
,
46
Andere PAH-verbundene Mutationen, die nichts mit der BMPR2-Signalgebung zu tun haben, beinhalten KCNK3 und CAV1. KCNK3 (Kaliumkanal-Unterfamilie K-Mitglied 3) codiert einen pH-empfindlichen Kaliumkanal, der den Gefäßtonus beeinflusst.
47
CAV1 (Caveolin-1) kodiert für ein Membranprotein, das für die Bildung von Lipidflößen oder Caveoli wesentlich ist, die zur Positionierung von BMP-Rezeptoren dienen.
48
Bis zu 25 % der sporadischen Fälle von pulmonaler kapillärer Hämangiomatose und pulmonaler venöser Verschlusskrankheit wurden Mutationen in EIF2AK4 (eukaryotischer Translationsinitiationsfaktor 2 Alpha-Kinase 4) zugeschrieben.
49
,
50
Neben Mutationen des Genoms standen auch epigenetische Mechanismen, die die Zellfunktion beeinflussen, im Fokus der neueren Forschung. Diese Mechanismen können DNA-Methylierung, Mikro-RNAs oder Modifikation von Histonproteinen umfassen, die die Expression von Wachstumsfaktorspiegeln oder Genexpression verändern und dadurch Zellwachstum und -proliferation beeinflussen.
51–54
Die Infiltration von Entzündungszellen wird in der Nähe von Bereichen des Gefäßumbaus bei PAH beobachtet, was das Interesse an der potenziellen Rolle von Zytokinen und Wachstumsfaktoren im Prozess der Gefäßerkrankung weckt. Es ist unklar, ob das Vorhandensein von Entzündungszellinfiltraten eine Folge von Hypoxie darstellt oder ob Entzündungsmediatoren eine Gefäßzellschädigung und -funktionsstörung fördern. Die Untersuchung vaskulärer Läsionen hat eine Invasion von Lymphozyten, Makrophagen, Mastzellen und dendritischen Zellen gezeigt.
55–57
Während in den Lungen von Patienten mit idiopathischer PAH ein Mangel an regulatorischen T-Zellen festgestellt wurde,
58
deuten eine Ausdehnung des ektopischen pulmonalen Lymphgewebes und das Vorhandensein von Autoantikörpern auf eine übermäßige B-Zell-Aktivierung hin.
59
Ektopisches Gewebe in der Nähe von Lungenarterien bei PAH kann mit dem Gefäßumbauprozess in Verbindung gebracht werden. Dendritische Zellen setzen Zytokine frei, die T- und B-Lymphozyten anziehen, ihr Überleben verbessern und zu einer entzündlichen Umgebung beitragen.
60
Makrophagen, die sich in der Nähe von Gefäßläsionen befinden, sind anfällig für die Wirkungen von IL-6, das von aktivierten Adventitia-Fibroblasten freigesetzt wird, und nehmen einen Phänotyp mit proinflammatorischen Eigenschaften an.
61
Makrophagen sind eine Quelle des aus Blutplättchen stammenden Wachstumsfaktors (PDGF), der ein potentes Mitogen und Chemoattraktor für Gefäßzellen ist.
62
Obwohl die genaue Rolle der Immunzellbiologie und der Entzündung bei der Pathogenese von PAH noch definiert werden muss, spricht das Vorhandensein ähnlicher pulmonaler Gefäßveränderungen sowohl bei PAH als auch bei einer signifikanten Anzahl von Patienten mit primären Autoimmunerkrankungen für eine Rolle von immunzellvermittelten Ereignissen in Umbau der Lungengefäße.Abgesehen von der Auswirkung genetischer und immunologischer oder entzündlicher Faktoren auf die strukturelle Umgestaltung gibt es Hinweise darauf, dass eine Endothelverletzung und -dysfunktion Ungleichgewichte in der Produktion von endogenen Mediatoren des Gefäßtonus, der Blutplättchenaggregation und der Zellproliferation verursachen. Immunhistochemische Studien haben signifikante Reduktionen der Stickoxid-Synthase- und Prostacyclin-Synthase-Spiegel im Lungengefäßendothel gezeigt, wo diese Enzyme eine entscheidende Rolle bei der Produktion von Stickoxid und Prostacyclin spielen, die beide gefäßerweiternde und antiproliferative Wirkungen auf Lungengefäßzellen haben.
63
,
64
Während die Produktion von Stickoxid und Prostacyclin durch pulmonalvaskuläres Endothel reduziert ist, ist die Produktion von Endothelin-1 durch Endothelzellen bei PAH erhöht.
65
Endothelin-1 fördert gegensätzliche Eigenschaften, einschließlich Vasokonstriktion und Zellproliferation. Endothelin-1, Survivin und vaskulärer endothelialer Wachstumsfaktor (VEGF) wurden in vaskulären plexiformen Läsionen isoliert und es wird angenommen, dass sie die Proliferation von Endothelzellen und glatten Muskelzellen verstärken, während sie die Apoptose hemmen.
65–67
Die Thromboxanproduktion durch das Lungenendothel wird ebenfalls erhöht, was zu einer Vasokonstriktion und einer In-situ-Thrombusbildung in den Lungenarterien führt.
68
Andere Faktoren mit potenzieller Rolle bei der Pathogenese von PAH sind Serotonin, Autoantikörper, dysfunktionale spannungsgesteuerte Kaliumkanäle und krebsähnliche Muster der Zellproliferation und Apoptoseresistenz. Serotonin kann die Vasokonstriktion und den Gefäßumbau fördern, indem es die Proliferation glatter Muskelzellen (SMC) und Fibroblasten stimuliert.
69–71
Die Expression von Anti-Endothelzellen- und Anti-Fibroblasten-Antikörpern gegen spezifische Zielantigene wurde bei idiopathischer und Sklerodermie-assoziierter PAH beobachtet, obwohl die Rolle dieser Autoimmuneffektoren bei der Pathogenese von PAH unklar ist.
72
,
73
Die Proliferation glatter Muskelzellen kann durch Serotonin-Transporter-Aktivierung des Thrombozyten-Wachstumsfaktor-beta (PDGF--Rezeptors stimuliert werden.
74
Intrazelluläres Calcium der glatten Muskulatur reguliert nicht nur die Kontraktion, sondern auch die Proliferation und den Widerstand gegen Apoptose. Herunterregulierung und Dysfunktion von spannungsgesteuerten Kaliumkanälen beeinflussen die Membranpolarisation und erhöhen den Calciumeinstrom, was wiederum die Kontraktion glatter Muskelzellen fördert und die Proliferation verstärkt, indem Zellen in den Zellzyklus getrieben werden.
39
,
75
,
76
In Lungengefäßendothelzellen, SMCs und Fibroblasten wurde eine Reihe von krebsähnlichen zellulären Verhaltensweisen beobachtet, darunter die monoklonale Expansion von Endothelzellen bei idiopathischer PAH, das Vorhandensein instabiler kurzer DNA-Mikrosatellitensequenzen in plexiformen Läsionen und ein anhaltender hyperproliferativer und apoptoseresistenter Zustand wenn Endothelzellen aus der in vivo-Umgebung entfernt werden.
77–79
Endothelzellen der Lungengefäße, SMCs und Fibroblasten sind für die Energieerzeugung stärker von der Glykolyse abhängig.
80–82
Mitochondrien wechseln ähnlich wie Krebszellen von der Glukoseoxidation zur ungekoppelten aeroben Glykolyse und fördern so die Bildung von Vorläufern für die DNA-Synthese und die schnelle Zellproliferation.
83
Der vaskuläre Umbau bei PAH unterscheidet sich von Krebs durch die Tatsache, dass sich pulmonale Gefäßzellen nicht ohne Kontrolle klonal vermehren.Pathophysiologie hinter pulmonaler Hypertonie in anderen WHO-GruppenObwohl angenommen wird, dass pulmonale Hypertonie in Nicht-PAH-WHO-Gruppen typischerweise durch Mechanismen entsteht, die für andere Krankheitsprozesse und ihre Auswirkungen auf den Lungenkreislauf spezifisch sind, gibt es Hinweise auf Ähnlichkeiten in der Pathophysiologie zwischen WHO-Gruppen, die auf Endothelschäden oder Dysfunktionen wie die in PAH.
35
Pulmonale Hypertonie bei Linksherzerkrankung (PH-LHD) wird als Folge der retrograden Übertragung eines erhöhten linksatrialen Drucks auf den Lungenkreislauf angesehen. Erhöhter linksatrialer Druck kann eine Folge einer systolischen oder diastolischen linksventrikulären Dysfunktion oder einer Herzklappenerkrankung sein. Bei einigen Patienten der WHO-Gruppe 2 kann sich in den distalen Pulmonalarterien und -venolen ein vaskulärer Umbau mit PAH-ähnlichen Merkmalen entwickeln und auch nach Korrektur der zugrunde liegenden Ursache der linksatrialen Hypertonie bestehen bleiben.
84
Als Beispiel wurde gezeigt, dass Patienten mit Herzklappenerkrankungen nach Herzklappenreparatur und Entfernung der Quelle des erhöhten postkapillären Drucks Anzeichen einer anhaltenden pulmonalen Hypertonie aufweisen.
85
Es wird angenommen, dass eine endotheliale Dysfunktion, die aus einem verlängerten retrograden Anstieg des pulmonalen Gefäßdrucks resultiert, viele der beobachteten Veränderungen in der Gefäßstruktur und -funktion vermittelt.
89
Erhöhte Endothelinspiegel im Plasma werden bei Patienten mit PH-LHD berichtet, und die Stickoxidproduktion ist bei Herzinsuffizienz reduziert.
87
,
90
Es ist bekannt, dass die pulmonale Vaskulopathie bei PH-LHD die Aktivierung von Fibroblasten, die Proliferation der vaskulären Intima und Media, die perivaskuläre Infiltration mit Entzündungszellen und die erhöhte Zytokin- und Wachstumsfaktorexpression umfasst, die alle den bei PAH beobachteten Merkmalen ähneln.
86–88
Diese Befunde korrelieren mit Beobachtungen einer Gefäßumgestaltung, die der bei PAH beobachteten ähnelt.Pulmonale Hypertonie der WHO-Gruppe 3 aufgrund einer Lungenerkrankung und Hypoxie wird oft als Folge einer hypoxischen Vasokonstriktion und des Verlusts kleiner Gefäße und Kapillaren angesehen. Obwohl dies zutrifft, wurde chronische Hypoxie auch mit der Aktivität mehrerer Mediatoren in Verbindung gebracht, die die Gefäßstruktur und -funktion ähnlich der bei PAH festgestellten Gefäßumgestaltung beeinflussen.
91
,
92
Zusätzlicher Sauerstoff kann hilfreich sein, um die hypoxische Vasokonstriktion in frühen Stadien umzukehren, aber wenn die Krankheit fortschreitet und die Hypoxie chronischer wird, wird der Gefäßumbau zu einer bedeutenderen Ursache für den steigenden pulmonalen Gefäßwiderstand. Wie bei PAH spielt die Endothelbiologie eine Schlüsselrolle bei der Reaktion der Lungengefäße auf Hypoxie und wird durch TGF-B1 und Hypoxie-induzierbarer Faktor 1-alpha (HIF-1a) Signalwege beeinflusst.
81
,
91
,
93
,
94
In Versuchsmodellen induziert chronische Hypoxie die Expression des vaskulären endothelialen Wachstumsfaktors A (VEGFA) und seines Rezeptors VEGFR2 über HIF-1a.
91
,
95
,
96
Hypoxie induziert auch die Expression von TGF-B1 und erhöht dadurch die Produktion von PDGF-B. Es wurde gezeigt, dass PDGF-B die VEGFA-Expression und damit die Hypoxie-induzierte Endothelproliferation fördert.
91
,
97
Chronische Hypoxie ist ein Stimulus für die Freisetzung von Serotonin, was wiederum die Vasokonstriktion stimuliert und die Endothelproliferation und die Hypertrophie der glatten Muskulatur fördert.
92
,
98
Es wird berichtet, dass Hypoxie die Stickoxidproduktion der Endothelzellen verringert und die Freisetzung von Endothelin erhöht.
92
,
99
Als klinisches Korrelat wurde eine beeinträchtigte Endothel-abhängige SMC-Relaxation bei Patienten mit COPD unterschiedlichen Schweregrades beobachtet.
100
Es besteht ein beträchtliches Interesse an der Beziehung zwischen Tabakrauchen und vaskulärem Umbau und pulmonaler Hypertonie bei chronischen Lungenerkrankungen. Das Rauchen an sich scheint mehrere Auswirkungen auf die Lungengefäßbiologie zu haben, einschließlich erhöhter VEGF-Expression, reduzierter Stickoxid-Synthase-Spiegel, erhöhter Infiltration von Entzündungszellen, Endothelhyperplasie und gestörter Mitochondrienfunktion.
101–103
Obwohl insgesamt kaum verstanden, ist klar, dass die Ursachen der pulmonalen Hypertonie bei Patienten mit chronischer Lungenerkrankung und Hypoxie viel tiefer gehen als hypoxische Vasokonstriktion und Verlust kleiner Gefäße.Die pulmonale Hypertonie ist eine bekannte Komplikation der akuten pulmonalen Thromboembolie. In der akuten Phase einer Thromboembolie kann der pulmonalvaskuläre Widerstand durch eine signifikante Gerinnselbelastung erhöht sein, die das pulmonalvaskuläre Bett verstopft. Typischerweise wird durch Thrombolyse und/oder Antikoagulation das obstruktive Gerinnsel beseitigt und der normale Gefäßwiderstand wiederhergestellt. Doch selbst bei kontinuierlicher Antikoagulation können Lungenperfusionsstörungen in über 50 % der Fälle länger als 3 Monate nach einem akuten Ereignis bestehen bleiben.
104
Ein kleiner Prozentsatz dieser Patienten entwickelt später eine chronisch thromboembolische pulmonale Hypertonie der WHO-Gruppe 4 (CTEPH). Studien schätzen die jährliche Inzidenz von CTEPH nach einer akuten Lungenembolie auf 0,4 % bis 6,2 %.
105
Die wahre Inzidenz ist jedoch ungewiss, da aktuelle Schätzungen der CTEPH-Inzidenz nach akuter LE Patienten umfassen können, die bereits vor dem akuten Ereignis eine chronische Thromboembolie hatten. CTEPH kann mehrere Monate oder Jahre nach kontinuierlicher Antikoagulation und ohne symptomatische rezidivierende akute Ereignisse diagnostiziert werden. Die pulmonale Hypertonie in dieser Population ist teilweise eine Folge eines sich nicht auflösenden Thrombus. Im Gegensatz zu dem charakteristischen frischen Gerinnsel bei akuter Lungenembolie besteht das chronisch flussbegrenzende Material bei CTEPH aus einem gelben fibrotischen Material, das fest an der Gefäßwand haftet und aus Kollagen, Elastin, Entzündungszellen und Rekanalisationsgefäßen besteht.
105
Es gibt mehrere vorgeschlagene Theorien, die sich auf die Nichtauflösung von thrombotischem Material bei Patienten beziehen, die CTEPH entwickeln. Wenn die thromboembolische Belastung groß ist, kann das intrinsische lytische System aufgrund unzureichender lytischer Kapazität oder Fähigkeit, die gesamte Gerinnselmasse zu erreichen, möglicherweise nicht in der Lage sein, eine vollständige Auflösung zu erreichen. Eine andere Möglichkeit besteht darin, dass Patienten, die behandelt werden, möglicherweise nicht ausreichend oder nicht lange genug antikoaguliert werden. Zugrunde liegende Autoimmunerkrankungen, Nicht-O-Blutgruppe, Splenektomie in der Vorgeschichte, ventrikulo-atriale Shunts, Schilddrüsenersatz und eine Vorgeschichte von Malignität waren in einer großen Studie, in der Patienten mit CTEPH mit Patienten mit anderen Formen der pulmonalen Hypertonie verglichen wurden, alle mit einem höheren CTEPH-Risiko verbunden.
106
Bei CTEPH wurde über höhere Konzentrationen mehrerer Entzündungsmarker und Entzündungsmediatoren berichtet, was darauf hindeutet, dass die zugrunde liegende Entzündung irgendwie beteiligt ist.
107
,
108
Mutationen, die abnormales Fibrinogen und damit eine abnormale Fibrinstruktur und Resistenz gegenüber Plasmin-vermittelter Lyse verursachen, wurden identifiziert.
109–111
Anomalien der Thrombozytenfunktion und dysfunktionale Angiogenese und Rekanalisation des Thrombus wurden ebenfalls vorgeschlagen, um die Nichtauflösung des Thrombus bei CTEPH zu erklären.
112
,
113
Die Nichtauflösung des Gerinnsels allein erklärt nicht die Entwicklung einer pulmonalen Hypertonie bei CTEPH. Ein Umbau kleiner Gefäße, ähnlich dem bei idiopathischer PAH beschriebenen, wird auch bei CTEPH beobachtet und betrifft Gefäße mit ungehindertem Fluss sowie solche, die distal von flussbegrenzenden Thromben liegen. Weiterhin erstrecken sich die Charakteristika der arteriellen Vaskulopathie dahingehend, Venolen und kleine Venen einzubeziehen. Die Entwicklung einer Vaskulopathie in unverstopften Arterien wurde als Folge eines umgeleiteten Flusses mit erhöhtem Druck und Scherbeanspruchung angesehen, was zu einer Endothelverletzung führt.
114
Dieser Mechanismus würde jedoch nicht die Entwicklung einer Arteriopathie distal zu obstruktiven Thromben erklären. Es wurden Anastamosen zwischen den Bronchial- und Pulmonalarterien identifiziert, die einen Fluss distal zu pulmonalarteriellen Obstruktionen ermöglichen können.
115
Es wird die Theorie aufgestellt, dass das Aussetzen kleiner Arterien distal des Thrombus gegenüber systemischem Druck den Gefäßumbau in diesen Arterien fördern kann. Abgesehen von diesen mechanischen Überlegungen gibt es Hinweise darauf, dass bei CTEPH auch molekulare Faktoren existieren, die den Gefäßumbau begünstigen. Es ist bekannt, dass die Spiegel des endogenen Vasodilatators Stickstoffmonoxid sowohl bei Patienten mit PAH als auch mit CTEPH reduziert sind.
116
Darüber hinaus sind die Spiegel des Stickstoffmonoxid-Synthase-Inhibitors, asymmetrisches Dimethylarginin, erhöht und können die günstigen gefäßerweiternden und antiproliferativen Wirkungen von Stickstoffmonoxid auf vaskulärer Ebene einschränken.
117
Erhöhte Endothelinspiegel wurden, wie bei PAH erwähnt, auch bei CTEPH-Patienten beobachtet.
118
Obwohl Thrombose das primäre Ereignis bei CTEPH ist, gibt es immer mehr Hinweise darauf, dass die Entwicklung von pulmonaler Hypertonie mit Abweichungen in einem komplexen molekularen Signalsystem einhergeht, das in vielerlei Hinsicht dem bei PAH beobachteten ähnelt.
Risikobewertung und Behandlung von PAH der WHO-Gruppe 1Die Behandlungsmöglichkeiten der pulmonalen arteriellen Hypertonie haben sich in den letzten Jahren rasant weiterentwickelt. Vor 1995 umfassten die Behandlungsoptionen für Patienten mit PAH Diuretika, Antikoagulation, Digoxin, Kalziumkanalblocker und Sauerstoff. Patienten mit ungehindertem Krankheitsverlauf könnten letztendlich eine Lungentransplantation benötigen. 1995 war Epoprostenol die erste zielgerichtete Therapie, die von der United States Federal Drug Administration für die Behandlung von PAH zugelassen wurde. Seitdem wurden mehrere zusätzliche zielgerichtete Therapien zugelassen, die die Zahl der nicht-chirurgischen Optionen für Patienten erweitern. Gegenwärtig verfügbare Behandlungsmittel für PAH zielen auf pathophysiologische Mechanismen in Stickoxid-, Endothelin- oder Prostacyclin-Wegen ab. Da immer mehr medizinische Therapien zur Verfügung stehen, Es gibt also Hinweise darauf, dass die Kombination von Wirkstoffen und die gleichzeitige Ausrichtung auf mehrere Signalwege oft vorteilhafter sind als deren einzelne Anwendung. Die Ausweitung des Einsatzes von zielgerichteten PAH-Therapien der WHO-Gruppe 1 zur Behandlung in anderen WHO-Gruppen war nur begrenzt erfolgreich, so dass es an guten Optionen für die Behandlung dieser Patienten mangelt. Daher wird sich der Fokus auf verfügbare Behandlungen in dieser Übersicht hauptsächlich auf Optionen für PAH der WHO-Gruppe 1 und CTEPH der Gruppe 4 beziehen, wobei neuere Untersuchungen zur Ausweitung der Mittel zur Verwendung in anderen WHO-Gruppen erwähnt werden.Die Auswahl aus den verfügbaren medizinischen Therapien zur Ausarbeitung eines wirksamen PAH-Behandlungsplans beinhaltet einen fortlaufenden Prozess der Risikobewertung und Ergebnisüberwachung. PAH ist eine fortschreitende Krankheit, selbst im gegenwärtigen Zeitalter der zielgerichteten Therapie, und eine sorgfältige Risiko- und Ergebnisüberwachung ist ein wesentlicher Bestandteil des Behandlungsprozesses. Die Beobachtung, dass es eine starke Korrelation zwischen dem Überleben und bestimmten klinischen Merkmalen bei PAH gibt, wie z. B. 6-Minuten-Gehstrecke, Funktionsklasse, hämodynamische Maße und andere Parameter, hat zur Entwicklung mehrerer Instrumente zur Risikobewertung geführt. 119 Die Funktion des rechten Ventrikels ist weithin als Determinante des Outcomes bei PAH anerkannt. Nichtinvasive Messungen der rechtsventrikulären Struktur und Funktion durch Echokardiographie oder Herz-MRT sind bei der Risikostratifizierung hilfreich und wurden in die Behandlungsleitlinien aufgenommen. 120 , 121 Die jüngsten Leitlinien der European Society of Cardiology/European Respiratory Society umfassen den Bereich des rechten Vorhofs, die systolische Exkursion in der Trikuspidalringebene/den systolischen Pulmonalarteriendruck (TAPSE/sPAP) und das Vorhandensein eines Perikardergusses unter den Risikostratifizierungsvariablen. 3 Risikobewertungs-Tools integrieren mehrere klinische Parameter, um vorherzusagen, ob ein Patient ein niedriges, mittleres oder hohes Risiko für einen kurzfristigen Tod durch PAH hat, und leiten somit den Ansatz für das medizinische Behandlungsdesign. Zwei der am häufigsten verwendeten Tools zur Risikobewertung sind der ESC/ERS-Risikobewertungsalgorithmus 3 und der REVEAL-Risikorechner. 122 Der REVEAL-Risikorechner wurde aus Daten erstellt, die im amerikanischen REVEAL-PAH-Register erfasst wurden, bei dem es sich um ein 3-jähriges Längsschnittregister von 2967 PAH-Patienten der WHO-Gruppe 1 mit gesammelten Daten zu klinischen Merkmalen, Bewertung, Behandlung und Ergebnissen handelte. 123 Eine validierte Überarbeitung des REVEAL-Rechners (REVEAL 2.0) enthält eine gekürzte Version (REVEAL Lite 2), die die Nützlichkeit in ambulanten Einrichtungen verbessert, in denen eine regelmäßige Nachsorge erfolgt. 124 , 125 Eine Risikobewertung wird in der Regel bei jedem regelmäßigen Nachsorgebesuch durchgeführt, und der Behandlungsplan wird nach Bedarf angepasst, um einen niedrigen Risikostatus zu erreichen und aufrechtzuerhalten. Basierend auf der REVEAL 2.0-Risikostratifizierung haben Patienten in der Gruppe mit niedrigem Risiko eine vorhergesagte 1-Jahres-Überlebensrate von > 94 %, eine 1-Jahres-Überlebensrate mit mittlerem Risiko von 70 bis < 94 % und eine 1-Jahres-Überlebensrate mit hohem Risiko von < 70 %. Aktuelle Behandlungsrichtlinien integrieren eine Risikobewertung in den Algorithmus, um Behandlungspläne zu ändern.Stickoxid-WegStickoxid (NO) ist ein endogener Vasodilatator, der vom Lungenendothel produziert wird. Stickoxidsynthase (NOS) wandelt L-Arginin in NO um, das auf benachbarte glatte Muskelzellen einwirkt, wo es die Umwandlung von Guanosintriphosphat (GTP) in zyklisches Guanosinmonophosphat (cGMP) durch Guanylatcyclase katalysiert. Zyklisches GMP wiederum fördert die Entspannung der glatten Muskulatur und Vasodilatation, reguliert aber auch Zellproliferation, Apoptose und Entzündung. 126 , 127 Zyklisches GMP wird durch endogene Phosphodiesterase-5 (PDE-5) neutralisiert, wodurch seine Wirkung begrenzt wird. Patienten mit PAH haben eine mangelhafte NOS-Aktivität in den Lungengefäßen, was zu einer geringen NO-Produktion führt. 63 Günstige Ergebnisse bei der Behandlung von PAH wurden erzielt, indem der NO-cGMP-Weg mit Wirkstoffen angegriffen wurde, die PDE-5 hemmen (PDE-5-Inhibitoren) oder Guanylatcyclase direkt stimulieren (lösliche Guanylatcyclase-Stimulatoren).Phosphodiesterase-5-Inhibitoren begrenzen die Inaktivierung von cGMP und verstärken dadurch seine Wirkung auf die glatte Gefäßmuskulatur. Drei PDE-5-Hemmer, Sildenafil, Tadalafil und Vardenafil , verbessern nachweislich die pulmonale Hämodynamik, die Belastungsfähigkeit und die Symptome bei Patienten mit PAH, einschließlich solcher mit Bindegewebserkrankungen. 128–130 Außerdem hat sich gezeigt, dass Tadalafil die Zeit bis zur klinischen Verschlechterung verlängert. 129 Diese Mittel können eine schwere Hypotonie auslösen, wenn sie zusammen mit Nitroglycerin verwendet werden, und daher ist die Verwendung von Nitroglycerin mit PDE-5-Hemmern kontraindiziert. Die Sicherheit bei Schwangeren wurde nicht nachgewiesen; in Tierversuchen wu
Die sich entwickelnden Management- und Behandlungsoptionen für Patienten mit pulmonaler Hypertonie: Aktuelle Erkenntnisse und Herausforderungen Zusammenfassung:Pulmonale Hypertonie kann sich als pulmonalarterienspezifischer Krankheitsprozess ohne erkennbare Ursache entwickeln oder im Zusammenhang mit anderen kardiopulmonalen und systemischen Erkrankungen auftreten. Die Weltgesundheitsorganisation (WHO) klassifiziert pulmonale hypertensive Erkrankungen auf der Grundlage der primären Mechanismen, die einen erhöhten pulmonalen Gefäßwiderstand verursachen. Eine wirksame Behandlung der pulmonalen Hypertonie beginnt mit der genauen Diagnose und Klassifizierung der Krankheit, um die geeignete Behandlung zu bestimmen. Die pulmonale arterielle Hypertonie (PAH) ist eine besonders herausfordernde Form der pulmonalen Hypertonie, da es sich um einen fortschreitenden, hyperproliferativen arteriellen Prozess handelt, der unbehandelt zu Rechtsherzversagen und Tod führt. In den letzten zwei Jahrzehnten Unser Verständnis der Pathobiologie und Genetik hinter PAH hat sich weiterentwickelt und zur Entwicklung mehrerer zielgerichteter Krankheitsmodifikatoren geführt, die die Hämodynamik und Lebensqualität verbessern. Effektive Risikomanagementstrategien und aggressivere Behandlungsprotokolle haben auch bessere Ergebnisse für Patienten mit PAH ermöglicht. Für Patienten, bei denen es unter medikamentöser Therapie zu einer fortschreitenden PAH kommt, bleibt die Lungentransplantation eine lebensrettende Option. Neuere Arbeiten waren auf die Entwicklung wirksamer Behandlungsstrategien für andere Formen von pulmonaler Hypertonie gerichtet, wie etwa chronisch thromboembolischer pulmonaler Hypertonie (CTEPH) und pulmonaler Hypertonie aufgrund anderer Lungen- oder Herzerkrankungen. Die Entdeckung neuer Krankheitswege und Modifikatoren, die den Lungenkreislauf beeinflussen, ist ein fortlaufendes Gebiet intensiver Forschung.
Schlüsselwörter: pulmonale arterielle Hypertonie, chronisch thromboembolische pulmonale Hypertonie, Stickoxidweg, Endothelin-Antagonismus, Prostacyclin-AnalogonEinführungPulmonale Hypertonie (PH) ist definiert als ein mittlerer Pulmonalarteriendruck (PAP) größer oder gleich 20 mmHg, gemessen durch Rechtsherzkatheter. 1 , 3 Mehrere Mechanismen hinter der Entwicklung eines erhöhten Lungenarteriendrucks haben dazu geführt, dass die Weltgesundheitsorganisation (WHO) pulmonale hypertensive Erkrankungen in fünf verschiedene Gruppen eingeteilt hat. 1 Eine der größten Herausforderungen bei der Annäherung an einen Patienten mit pulmonaler Hypertonie ist die Aufgabe, die Krankheit richtig zu diagnostizieren und zu klassifizieren, um die geeignete Behandlung zu bestimmen. In einigen Fällen ist die optimale Behandlung einer assoziierten Erkrankung die primäre Behandlung der Wahl, aber in anderen Situationen können gezielte Medikamente oder chirurgische Maßnahmen indiziert sein. In den letzten zwei Jahrzehnten wurden erhebliche wissenschaftliche Anstrengungen unternommen, um die Pathobiologie hinter der pulmonalen arteriellen Hypertonie (PAH) der WHO-Gruppe 1 zu verstehen. PAH ist eine Erkrankung, die die Lungenarterien betrifft und durch spezifische Veränderungen in der Arterienmorphologie und einen steigenden Lungengefäßwiderstand gekennzeichnet ist, die zu Rechtsherzversagen und Tod führen können. Pathophysiologische Wege, die an PAH beteiligt sind, wurden definiert. 2 , 3 Eine Reihe neuer Mechanismen und Mediatoren werden derzeit untersucht. Die Entdeckung spezifischer krankheitsverursachender Mechanismen hat die Entwicklung zielgerichteter Therapien gefördert, die bei der Kontrolle der fortschreitenden Arteriopathie und der Symptome von PAH wirksam sind. Mehrere Untersuchungen sind im Gange, um zu klären, ob dieselben zielgerichteten Modifikatoren bei der Behandlung von pulmonaler Hypertonie in anderen WHO-Gruppen von Vorteil sein könnten. Diese Übersichtsarbeit beschreibt die grundlegenden Mechanismen, die an der Klassifizierung von pulmonal-hypertensiven Erkrankungen beteiligt sind, geht detaillierter auf das aktuelle Verständnis der PAH-Pathobiologie ein, skizziert aktuelle Managementstrategien, um optimale Ergebnisse für Patienten mit PAH zu erzielen, und fasst mehr zusammen jüngsten Bemühungen, Lösungen für die Behandlung von PH in anderen WHO-Gruppen zu finden.
Diagnose und Klassifizierung von pulmonalen hypertensiven ErkrankungenDie pulmonale Hypertonie wurde erstmals 1973 während des 1. Weltsymposiums über pulmonale Hypertonie in Genf, Schweiz, von der Weltgesundheitsorganisation nach pathologischen und klinischen Merkmalen klassifiziert. 4 Das Klassifizierungssystem der WHO wurde im Laufe der Zeit weiter verfeinert, wobei die jüngsten Aktualisierungen vom 6. Weltsymposium im Jahr 2018 und den Leitlinien der ESC/ERS (European Society of Cardiology/European Respiratory Society) von 2022 für die Diagnose und Behandlung von pulmonaler Hypertonie stammen. 1 , 3 Die Klassifizierung basiert auf den primären Mechanismen, die einen erhöhten pulmonalarteriellen Druck verursachen ( Abbildung 1 ). Patienten mit PAH der WHO-Gruppe 1 haben eine ausgeprägte Arteriopathie, die durch übermäßige Proliferation der zellulären Komponenten der Gefäßwand, Hypertrophie der glatten Muskulatur, In-situ-Thrombose und Bildung plexiformer Läsionen gekennzeichnet ist, die das Gefäßlumen verschließen. 5 , 6 Hyperproliferative Veränderungen, die in dieser Gruppe auftreten, können sich auch in pulmonalvenösen Strukturen entwickeln. PH der WHO-Gruppe 2 resultiert in erster Linie aus steigenden postkapillären Drücken, wie sie bei Erkrankungen beobachtet werden, die die linke Herzseite betreffen, wie Kardiomyopathien oder Herzklappenerkrankungen. Ein dritter Mechanismus, der pulmonale Hypertonie verursacht, wird durch Hypoxie und damit verbundene Vasokonstriktion vermittelt. Hypoxische Vasokonstriktion in der WHO-Gruppe 3 PH kann mit großer Höhe, Schlafapnoe und anderen Lungenerkrankungen wie Lungenfibrose oder Emphysem einhergehen. Eine Kreislaufobstruktion, die aus thromboembolischen oder anderen embolischen Ereignissen resultiert, kann zu einer pulmonalen Hypertonie der WHO-Gruppe 4 führen. Die endgültige Klassifikation, WHO-Gruppe 5, besteht aus Erkrankungen, die mit pulmonaler Hypertonie assoziiert sind, ohne einheitliche mechanistische Merkmale.

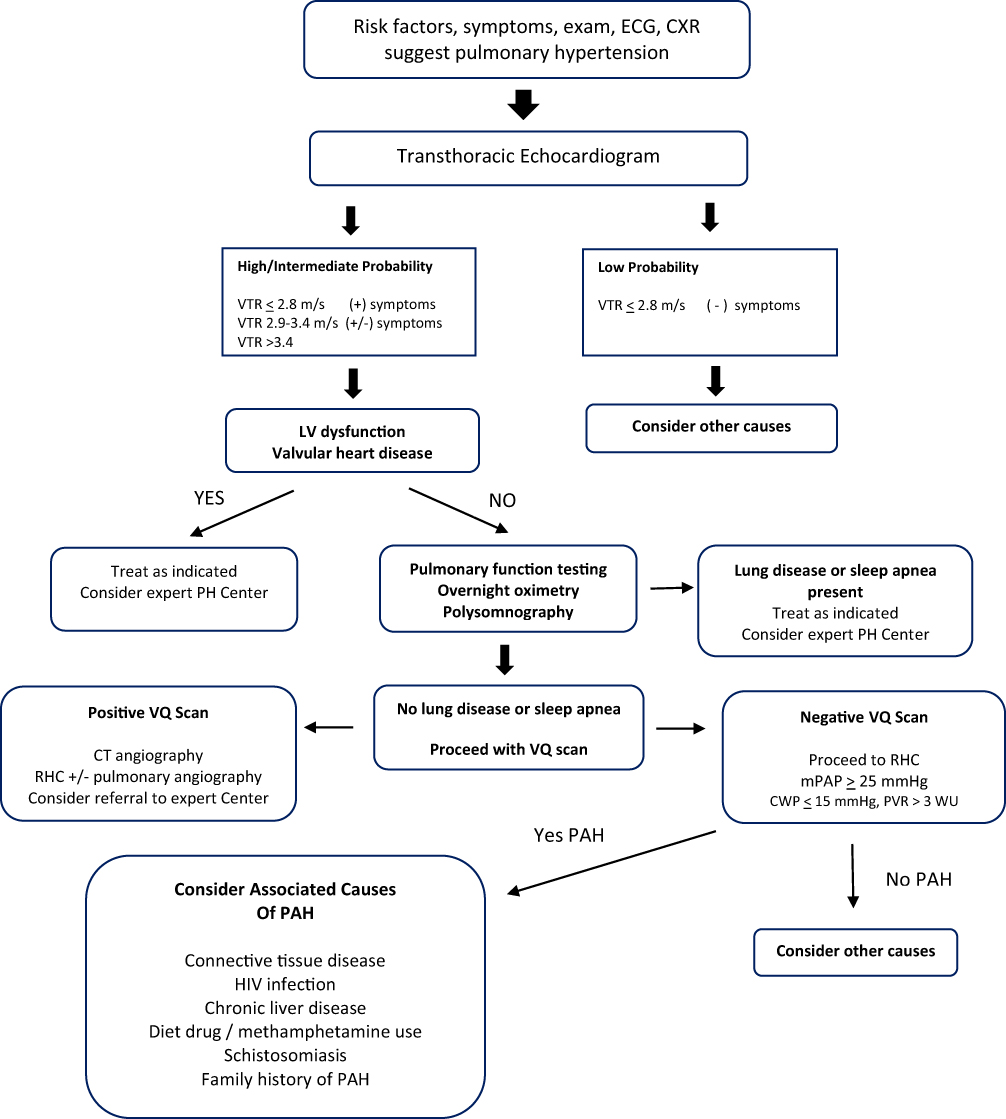

Pathophysiologie der pulmonalen arteriellen Hypertonie (WHO-Gruppe 1)Patienten, die als PAH der WHO-Gruppe 1 klassifiziert sind, teilen eine Vaskulopathie der kleinen präkapillaren Arterien, die durch übermäßige Endothelproliferation, Proliferation und Hypertrophie der glatten Muskulatur, In-situ-Thrombose und Bildung von vaskulären plexiformen Läsionen gekennzeichnet ist. 5 , 34 Diese vaskulären Veränderungen können auch in den postkapillaren Gefäßen bei bestimmten Erkrankungen wie systemischer Sklerose und pulmonaler Venenverschlusskrankheit gefunden werden. Neben der charakteristischen Umgestaltung des Lungengefäßsystems kommt es zu einem Verlust präkapillarer Arterien und einer übertriebenen Infiltration des perivaskulären Raums mit Entzündungszellen. 2 Die Pathogenese dieser Veränderungen ist ein komplexer Prozess mit Hinweisen auf genetische Faktoren, Zytokine und Wachstumsfaktoren, Ionenkanaldysfunktion und Gefäßverletzung mit endothelialer Dysfunktion, die zu einem Ungleichgewicht der endogenen vasomotorischen Regulation und der Zellproliferation im pulmonalen Gefäßbett führt. 2 , 35 , 36 Eine genetische Grundlage für PAH wurde erstmals im Jahr 2000 mit der Entdeckung einer Genmutation des knochenmorphogenetischen Proteinrezeptors II (BMPR2) bei Patienten mit vererbbarer PAH aufgedeckt. 37 , 38 Diese Mutation wird bei 70–80 % der Patienten mit erblicher PAH und bei 15–25 % der Patienten mit idiopathischer PAH identifiziert. 39 , 40 BMPR2 ist ein Mitglied der Gen-Superfamilie des transformierenden Wachstumsfaktors Beta (TGF-
und dient dazu, die Proliferation glatter Muskelzellen und die Apoptose von Endothelzellen zu begrenzen.
41
Sequenzvariationen in Genen, die für BMPR2-verwandte nachgeschaltete SMAD (Suppressor of Mothers Against Decapentaplegic)-Signaltransduktoren (SMAD1, SMAD4, SMAD8, SMAD9) codieren, wurden ebenfalls mit der Pathogenese von PAH in Verbindung gebracht.
42–44
Mutationen, an denen Activin-Rezeptor-ähnliche Kinase 1 (ACVRL1 oder ALK1) und Endoglin (ENG) beteiligt sind, machen 80 % der Fälle von hereditärer hämorrhagischer Teleangiektasie (HHT) aus, wobei 15–20 % dieser Patienten auch eine pulmonale Hypertonie entwickeln.
45
,
46
Andere PAH-verbundene Mutationen, die nichts mit der BMPR2-Signalgebung zu tun haben, beinhalten KCNK3 und CAV1. KCNK3 (Kaliumkanal-Unterfamilie K-Mitglied 3) codiert einen pH-empfindlichen Kaliumkanal, der den Gefäßtonus beeinflusst.
47
CAV1 (Caveolin-1) kodiert für ein Membranprotein, das für die Bildung von Lipidflößen oder Caveoli wesentlich ist, die zur Positionierung von BMP-Rezeptoren dienen.
48
Bis zu 25 % der sporadischen Fälle von pulmonaler kapillärer Hämangiomatose und pulmonaler venöser Verschlusskrankheit wurden Mutationen in EIF2AK4 (eukaryotischer Translationsinitiationsfaktor 2 Alpha-Kinase 4) zugeschrieben.
49
,
50
Neben Mutationen des Genoms standen auch epigenetische Mechanismen, die die Zellfunktion beeinflussen, im Fokus der neueren Forschung. Diese Mechanismen können DNA-Methylierung, Mikro-RNAs oder Modifikation von Histonproteinen umfassen, die die Expression von Wachstumsfaktorspiegeln oder Genexpression verändern und dadurch Zellwachstum und -proliferation beeinflussen.
51–54
Die Infiltration von Entzündungszellen wird in der Nähe von Bereichen des Gefäßumbaus bei PAH beobachtet, was das Interesse an der potenziellen Rolle von Zytokinen und Wachstumsfaktoren im Prozess der Gefäßerkrankung weckt. Es ist unklar, ob das Vorhandensein von Entzündungszellinfiltraten eine Folge von Hypoxie darstellt oder ob Entzündungsmediatoren eine Gefäßzellschädigung und -funktionsstörung fördern. Die Untersuchung vaskulärer Läsionen hat eine Invasion von Lymphozyten, Makrophagen, Mastzellen und dendritischen Zellen gezeigt.
55–57
Während in den Lungen von Patienten mit idiopathischer PAH ein Mangel an regulatorischen T-Zellen festgestellt wurde,
58
deuten eine Ausdehnung des ektopischen pulmonalen Lymphgewebes und das Vorhandensein von Autoantikörpern auf eine übermäßige B-Zell-Aktivierung hin.
59
Ektopisches Gewebe in der Nähe von Lungenarterien bei PAH kann mit dem Gefäßumbauprozess in Verbindung gebracht werden. Dendritische Zellen setzen Zytokine frei, die T- und B-Lymphozyten anziehen, ihr Überleben verbessern und zu einer entzündlichen Umgebung beitragen.
60
Makrophagen, die sich in der Nähe von Gefäßläsionen befinden, sind anfällig für die Wirkungen von IL-6, das von aktivierten Adventitia-Fibroblasten freigesetzt wird, und nehmen einen Phänotyp mit proinflammatorischen Eigenschaften an.
61
Makrophagen sind eine Quelle des aus Blutplättchen stammenden Wachstumsfaktors (PDGF), der ein potentes Mitogen und Chemoattraktor für Gefäßzellen ist.
62
Obwohl die genaue Rolle der Immunzellbiologie und der Entzündung bei der Pathogenese von PAH noch definiert werden muss, spricht das Vorhandensein ähnlicher pulmonaler Gefäßveränderungen sowohl bei PAH als auch bei einer signifikanten Anzahl von Patienten mit primären Autoimmunerkrankungen für eine Rolle von immunzellvermittelten Ereignissen in Umbau der Lungengefäße.Abgesehen von der Auswirkung genetischer und immunologischer oder entzündlicher Faktoren auf die strukturelle Umgestaltung gibt es Hinweise darauf, dass eine Endothelverletzung und -dysfunktion Ungleichgewichte in der Produktion von endogenen Mediatoren des Gefäßtonus, der Blutplättchenaggregation und der Zellproliferation verursachen. Immunhistochemische Studien haben signifikante Reduktionen der Stickoxid-Synthase- und Prostacyclin-Synthase-Spiegel im Lungengefäßendothel gezeigt, wo diese Enzyme eine entscheidende Rolle bei der Produktion von Stickoxid und Prostacyclin spielen, die beide gefäßerweiternde und antiproliferative Wirkungen auf Lungengefäßzellen haben.
63
,
64
Während die Produktion von Stickoxid und Prostacyclin durch pulmonalvaskuläres Endothel reduziert ist, ist die Produktion von Endothelin-1 durch Endothelzellen bei PAH erhöht.
65
Endothelin-1 fördert gegensätzliche Eigenschaften, einschließlich Vasokonstriktion und Zellproliferation. Endothelin-1, Survivin und vaskulärer endothelialer Wachstumsfaktor (VEGF) wurden in vaskulären plexiformen Läsionen isoliert und es wird angenommen, dass sie die Proliferation von Endothelzellen und glatten Muskelzellen verstärken, während sie die Apoptose hemmen.
65–67
Die Thromboxanproduktion durch das Lungenendothel wird ebenfalls erhöht, was zu einer Vasokonstriktion und einer In-situ-Thrombusbildung in den Lungenarterien führt.
68
Andere Faktoren mit potenzieller Rolle bei der Pathogenese von PAH sind Serotonin, Autoantikörper, dysfunktionale spannungsgesteuerte Kaliumkanäle und krebsähnliche Muster der Zellproliferation und Apoptoseresistenz. Serotonin kann die Vasokonstriktion und den Gefäßumbau fördern, indem es die Proliferation glatter Muskelzellen (SMC) und Fibroblasten stimuliert.
69–71
Die Expression von Anti-Endothelzellen- und Anti-Fibroblasten-Antikörpern gegen spezifische Zielantigene wurde bei idiopathischer und Sklerodermie-assoziierter PAH beobachtet, obwohl die Rolle dieser Autoimmuneffektoren bei der Pathogenese von PAH unklar ist.
72
,
73
Die Proliferation glatter Muskelzellen kann durch Serotonin-Transporter-Aktivierung des Thrombozyten-Wachstumsfaktor-beta (PDGF--Rezeptors stimuliert werden.
74
Intrazelluläres Calcium der glatten Muskulatur reguliert nicht nur die Kontraktion, sondern auch die Proliferation und den Widerstand gegen Apoptose. Herunterregulierung und Dysfunktion von spannungsgesteuerten Kaliumkanälen beeinflussen die Membranpolarisation und erhöhen den Calciumeinstrom, was wiederum die Kontraktion glatter Muskelzellen fördert und die Proliferation verstärkt, indem Zellen in den Zellzyklus getrieben werden.
39
,
75
,
76
In Lungengefäßendothelzellen, SMCs und Fibroblasten wurde eine Reihe von krebsähnlichen zellulären Verhaltensweisen beobachtet, darunter die monoklonale Expansion von Endothelzellen bei idiopathischer PAH, das Vorhandensein instabiler kurzer DNA-Mikrosatellitensequenzen in plexiformen Läsionen und ein anhaltender hyperproliferativer und apoptoseresistenter Zustand wenn Endothelzellen aus der in vivo-Umgebung entfernt werden.
77–79
Endothelzellen der Lungengefäße, SMCs und Fibroblasten sind für die Energieerzeugung stärker von der Glykolyse abhängig.
80–82
Mitochondrien wechseln ähnlich wie Krebszellen von der Glukoseoxidation zur ungekoppelten aeroben Glykolyse und fördern so die Bildung von Vorläufern für die DNA-Synthese und die schnelle Zellproliferation.
83
Der vaskuläre Umbau bei PAH unterscheidet sich von Krebs durch die Tatsache, dass sich pulmonale Gefäßzellen nicht ohne Kontrolle klonal vermehren.Pathophysiologie hinter pulmonaler Hypertonie in anderen WHO-GruppenObwohl angenommen wird, dass pulmonale Hypertonie in Nicht-PAH-WHO-Gruppen typischerweise durch Mechanismen entsteht, die für andere Krankheitsprozesse und ihre Auswirkungen auf den Lungenkreislauf spezifisch sind, gibt es Hinweise auf Ähnlichkeiten in der Pathophysiologie zwischen WHO-Gruppen, die auf Endothelschäden oder Dysfunktionen wie die in PAH.
35
Pulmonale Hypertonie bei Linksherzerkrankung (PH-LHD) wird als Folge der retrograden Übertragung eines erhöhten linksatrialen Drucks auf den Lungenkreislauf angesehen. Erhöhter linksatrialer Druck kann eine Folge einer systolischen oder diastolischen linksventrikulären Dysfunktion oder einer Herzklappenerkrankung sein. Bei einigen Patienten der WHO-Gruppe 2 kann sich in den distalen Pulmonalarterien und -venolen ein vaskulärer Umbau mit PAH-ähnlichen Merkmalen entwickeln und auch nach Korrektur der zugrunde liegenden Ursache der linksatrialen Hypertonie bestehen bleiben.
84
Als Beispiel wurde gezeigt, dass Patienten mit Herzklappenerkrankungen nach Herzklappenreparatur und Entfernung der Quelle des erhöhten postkapillären Drucks Anzeichen einer anhaltenden pulmonalen Hypertonie aufweisen.
85
Es wird angenommen, dass eine endotheliale Dysfunktion, die aus einem verlängerten retrograden Anstieg des pulmonalen Gefäßdrucks resultiert, viele der beobachteten Veränderungen in der Gefäßstruktur und -funktion vermittelt.
89
Erhöhte Endothelinspiegel im Plasma werden bei Patienten mit PH-LHD berichtet, und die Stickoxidproduktion ist bei Herzinsuffizienz reduziert.
87
,
90
Es ist bekannt, dass die pulmonale Vaskulopathie bei PH-LHD die Aktivierung von Fibroblasten, die Proliferation der vaskulären Intima und Media, die perivaskuläre Infiltration mit Entzündungszellen und die erhöhte Zytokin- und Wachstumsfaktorexpression umfasst, die alle den bei PAH beobachteten Merkmalen ähneln.
86–88
Diese Befunde korrelieren mit Beobachtungen einer Gefäßumgestaltung, die der bei PAH beobachteten ähnelt.Pulmonale Hypertonie der WHO-Gruppe 3 aufgrund einer Lungenerkrankung und Hypoxie wird oft als Folge einer hypoxischen Vasokonstriktion und des Verlusts kleiner Gefäße und Kapillaren angesehen. Obwohl dies zutrifft, wurde chronische Hypoxie auch mit der Aktivität mehrerer Mediatoren in Verbindung gebracht, die die Gefäßstruktur und -funktion ähnlich der bei PAH festgestellten Gefäßumgestaltung beeinflussen.
91
,
92
Zusätzlicher Sauerstoff kann hilfreich sein, um die hypoxische Vasokonstriktion in frühen Stadien umzukehren, aber wenn die Krankheit fortschreitet und die Hypoxie chronischer wird, wird der Gefäßumbau zu einer bedeutenderen Ursache für den steigenden pulmonalen Gefäßwiderstand. Wie bei PAH spielt die Endothelbiologie eine Schlüsselrolle bei der Reaktion der Lungengefäße auf Hypoxie und wird durch TGF-B1 und Hypoxie-induzierbarer Faktor 1-alpha (HIF-1a) Signalwege beeinflusst.
81
,
91
,
93
,
94
In Versuchsmodellen induziert chronische Hypoxie die Expression des vaskulären endothelialen Wachstumsfaktors A (VEGFA) und seines Rezeptors VEGFR2 über HIF-1a.
91
,
95
,
96
Hypoxie induziert auch die Expression von TGF-B1 und erhöht dadurch die Produktion von PDGF-B. Es wurde gezeigt, dass PDGF-B die VEGFA-Expression und damit die Hypoxie-induzierte Endothelproliferation fördert.
91
,
97
Chronische Hypoxie ist ein Stimulus für die Freisetzung von Serotonin, was wiederum die Vasokonstriktion stimuliert und die Endothelproliferation und die Hypertrophie der glatten Muskulatur fördert.
92
,
98
Es wird berichtet, dass Hypoxie die Stickoxidproduktion der Endothelzellen verringert und die Freisetzung von Endothelin erhöht.
92
,
99
Als klinisches Korrelat wurde eine beeinträchtigte Endothel-abhängige SMC-Relaxation bei Patienten mit COPD unterschiedlichen Schweregrades beobachtet.
100
Es besteht ein beträchtliches Interesse an der Beziehung zwischen Tabakrauchen und vaskulärem Umbau und pulmonaler Hypertonie bei chronischen Lungenerkrankungen. Das Rauchen an sich scheint mehrere Auswirkungen auf die Lungengefäßbiologie zu haben, einschließlich erhöhter VEGF-Expression, reduzierter Stickoxid-Synthase-Spiegel, erhöhter Infiltration von Entzündungszellen, Endothelhyperplasie und gestörter Mitochondrienfunktion.
101–103
Obwohl insgesamt kaum verstanden, ist klar, dass die Ursachen der pulmonalen Hypertonie bei Patienten mit chronischer Lungenerkrankung und Hypoxie viel tiefer gehen als hypoxische Vasokonstriktion und Verlust kleiner Gefäße.Die pulmonale Hypertonie ist eine bekannte Komplikation der akuten pulmonalen Thromboembolie. In der akuten Phase einer Thromboembolie kann der pulmonalvaskuläre Widerstand durch eine signifikante Gerinnselbelastung erhöht sein, die das pulmonalvaskuläre Bett verstopft. Typischerweise wird durch Thrombolyse und/oder Antikoagulation das obstruktive Gerinnsel beseitigt und der normale Gefäßwiderstand wiederhergestellt. Doch selbst bei kontinuierlicher Antikoagulation können Lungenperfusionsstörungen in über 50 % der Fälle länger als 3 Monate nach einem akuten Ereignis bestehen bleiben.
104
Ein kleiner Prozentsatz dieser Patienten entwickelt später eine chronisch thromboembolische pulmonale Hypertonie der WHO-Gruppe 4 (CTEPH). Studien schätzen die jährliche Inzidenz von CTEPH nach einer akuten Lungenembolie auf 0,4 % bis 6,2 %.
105
Die wahre Inzidenz ist jedoch ungewiss, da aktuelle Schätzungen der CTEPH-Inzidenz nach akuter LE Patienten umfassen können, die bereits vor dem akuten Ereignis eine chronische Thromboembolie hatten. CTEPH kann mehrere Monate oder Jahre nach kontinuierlicher Antikoagulation und ohne symptomatische rezidivierende akute Ereignisse diagnostiziert werden. Die pulmonale Hypertonie in dieser Population ist teilweise eine Folge eines sich nicht auflösenden Thrombus. Im Gegensatz zu dem charakteristischen frischen Gerinnsel bei akuter Lungenembolie besteht das chronisch flussbegrenzende Material bei CTEPH aus einem gelben fibrotischen Material, das fest an der Gefäßwand haftet und aus Kollagen, Elastin, Entzündungszellen und Rekanalisationsgefäßen besteht.
105
Es gibt mehrere vorgeschlagene Theorien, die sich auf die Nichtauflösung von thrombotischem Material bei Patienten beziehen, die CTEPH entwickeln. Wenn die thromboembolische Belastung groß ist, kann das intrinsische lytische System aufgrund unzureichender lytischer Kapazität oder Fähigkeit, die gesamte Gerinnselmasse zu erreichen, möglicherweise nicht in der Lage sein, eine vollständige Auflösung zu erreichen. Eine andere Möglichkeit besteht darin, dass Patienten, die behandelt werden, möglicherweise nicht ausreichend oder nicht lange genug antikoaguliert werden. Zugrunde liegende Autoimmunerkrankungen, Nicht-O-Blutgruppe, Splenektomie in der Vorgeschichte, ventrikulo-atriale Shunts, Schilddrüsenersatz und eine Vorgeschichte von Malignität waren in einer großen Studie, in der Patienten mit CTEPH mit Patienten mit anderen Formen der pulmonalen Hypertonie verglichen wurden, alle mit einem höheren CTEPH-Risiko verbunden.
106
Bei CTEPH wurde über höhere Konzentrationen mehrerer Entzündungsmarker und Entzündungsmediatoren berichtet, was darauf hindeutet, dass die zugrunde liegende Entzündung irgendwie beteiligt ist.
107
,
108
Mutationen, die abnormales Fibrinogen und damit eine abnormale Fibrinstruktur und Resistenz gegenüber Plasmin-vermittelter Lyse verursachen, wurden identifiziert.
109–111
Anomalien der Thrombozytenfunktion und dysfunktionale Angiogenese und Rekanalisation des Thrombus wurden ebenfalls vorgeschlagen, um die Nichtauflösung des Thrombus bei CTEPH zu erklären.
112
,
113
Die Nichtauflösung des Gerinnsels allein erklärt nicht die Entwicklung einer pulmonalen Hypertonie bei CTEPH. Ein Umbau kleiner Gefäße, ähnlich dem bei idiopathischer PAH beschriebenen, wird auch bei CTEPH beobachtet und betrifft Gefäße mit ungehindertem Fluss sowie solche, die distal von flussbegrenzenden Thromben liegen. Weiterhin erstrecken sich die Charakteristika der arteriellen Vaskulopathie dahingehend, Venolen und kleine Venen einzubeziehen. Die Entwicklung einer Vaskulopathie in unverstopften Arterien wurde als Folge eines umgeleiteten Flusses mit erhöhtem Druck und Scherbeanspruchung angesehen, was zu einer Endothelverletzung führt.
114
Dieser Mechanismus würde jedoch nicht die Entwicklung einer Arteriopathie distal zu obstruktiven Thromben erklären. Es wurden Anastamosen zwischen den Bronchial- und Pulmonalarterien identifiziert, die einen Fluss distal zu pulmonalarteriellen Obstruktionen ermöglichen können.
115
Es wird die Theorie aufgestellt, dass das Aussetzen kleiner Arterien distal des Thrombus gegenüber systemischem Druck den Gefäßumbau in diesen Arterien fördern kann. Abgesehen von diesen mechanischen Überlegungen gibt es Hinweise darauf, dass bei CTEPH auch molekulare Faktoren existieren, die den Gefäßumbau begünstigen. Es ist bekannt, dass die Spiegel des endogenen Vasodilatators Stickstoffmonoxid sowohl bei Patienten mit PAH als auch mit CTEPH reduziert sind.
116
Darüber hinaus sind die Spiegel des Stickstoffmonoxid-Synthase-Inhibitors, asymmetrisches Dimethylarginin, erhöht und können die günstigen gefäßerweiternden und antiproliferativen Wirkungen von Stickstoffmonoxid auf vaskulärer Ebene einschränken.
117
Erhöhte Endothelinspiegel wurden, wie bei PAH erwähnt, auch bei CTEPH-Patienten beobachtet.
118
Obwohl Thrombose das primäre Ereignis bei CTEPH ist, gibt es immer mehr Hinweise darauf, dass die Entwicklung von pulmonaler Hypertonie mit Abweichungen in einem komplexen molekularen Signalsystem einhergeht, das in vielerlei Hinsicht dem bei PAH beobachteten ähnelt.Risikobewertung und Behandlung von PAH der WHO-Gruppe 1Die Behandlungsmöglichkeiten der pulmonalen arteriellen Hypertonie haben sich in den letzten Jahren rasant weiterentwickelt. Vor 1995 umfassten die Behandlungsoptionen für Patienten mit PAH Diuretika, Antikoagulation, Digoxin, Kalziumkanalblocker und Sauerstoff. Patienten mit ungehindertem Krankheitsverlauf könnten letztendlich eine Lungentransplantation benötigen. 1995 war Epoprostenol die erste zielgerichtete Therapie, die von der United States Federal Drug Administration für die Behandlung von PAH zugelassen wurde. Seitdem wurden mehrere zusätzliche zielgerichtete Therapien zugelassen, die die Zahl der nicht-chirurgischen Optionen für Patienten erweitern. Gegenwärtig verfügbare Behandlungsmittel für PAH zielen auf pathophysiologische Mechanismen in Stickoxid-, Endothelin- oder Prostacyclin-Wegen ab. Da immer mehr medizinische Therapien zur Verfügung stehen, Es gibt also Hinweise darauf, dass die Kombination von Wirkstoffen und die gleichzeitige Ausrichtung auf mehrere Signalwege oft vorteilhafter sind als deren einzelne Anwendung. Die Ausweitung des Einsatzes von zielgerichteten PAH-Therapien der WHO-Gruppe 1 zur Behandlung in anderen WHO-Gruppen war nur begrenzt erfolgreich, so dass es an guten Optionen für die Behandlung dieser Patienten mangelt. Daher wird sich der Fokus auf verfügbare Behandlungen in dieser Übersicht hauptsächlich auf Optionen für PAH der WHO-Gruppe 1 und CTEPH der Gruppe 4 beziehen, wobei neuere Untersuchungen zur Ausweitung der Mittel zur Verwendung in anderen WHO-Gruppen erwähnt werden.Die Auswahl aus den verfügbaren medizinischen Therapien zur Ausarbeitung eines wirksamen PAH-Behandlungsplans beinhaltet einen fortlaufenden Prozess der Risikobewertung und Ergebnisüberwachung. PAH ist eine fortschreitende Krankheit, selbst im gegenwärtigen Zeitalter der zielgerichteten Therapie, und eine sorgfältige Risiko- und Ergebnisüberwachung ist ein wesentlicher Bestandteil des Behandlungsprozesses. Die Beobachtung, dass es eine starke Korrelation zwischen dem Überleben und bestimmten klinischen Merkmalen bei PAH gibt, wie z. B. 6-Minuten-Gehstrecke, Funktionsklasse, hämodynamische Maße und andere Parameter, hat zur Entwicklung mehrerer Instrumente zur Risikobewertung geführt. 119 Die Funktion des rechten Ventrikels ist weithin als Determinante des Outcomes bei PAH anerkannt. Nichtinvasive Messungen der rechtsventrikulären Struktur und Funktion durch Echokardiographie oder Herz-MRT sind bei der Risikostratifizierung hilfreich und wurden in die Behandlungsleitlinien aufgenommen. 120 , 121 Die jüngsten Leitlinien der European Society of Cardiology/European Respiratory Society umfassen den Bereich des rechten Vorhofs, die systolische Exkursion in der Trikuspidalringebene/den systolischen Pulmonalarteriendruck (TAPSE/sPAP) und das Vorhandensein eines Perikardergusses unter den Risikostratifizierungsvariablen. 3 Risikobewertungs-Tools integrieren mehrere klinische Parameter, um vorherzusagen, ob ein Patient ein niedriges, mittleres oder hohes Risiko für einen kurzfristigen Tod durch PAH hat, und leiten somit den Ansatz für das medizinische Behandlungsdesign. Zwei der am häufigsten verwendeten Tools zur Risikobewertung sind der ESC/ERS-Risikobewertungsalgorithmus 3 und der REVEAL-Risikorechner. 122 Der REVEAL-Risikorechner wurde aus Daten erstellt, die im amerikanischen REVEAL-PAH-Register erfasst wurden, bei dem es sich um ein 3-jähriges Längsschnittregister von 2967 PAH-Patienten der WHO-Gruppe 1 mit gesammelten Daten zu klinischen Merkmalen, Bewertung, Behandlung und Ergebnissen handelte. 123 Eine validierte Überarbeitung des REVEAL-Rechners (REVEAL 2.0) enthält eine gekürzte Version (REVEAL Lite 2), die die Nützlichkeit in ambulanten Einrichtungen verbessert, in denen eine regelmäßige Nachsorge erfolgt. 124 , 125 Eine Risikobewertung wird in der Regel bei jedem regelmäßigen Nachsorgebesuch durchgeführt, und der Behandlungsplan wird nach Bedarf angepasst, um einen niedrigen Risikostatus zu erreichen und aufrechtzuerhalten. Basierend auf der REVEAL 2.0-Risikostratifizierung haben Patienten in der Gruppe mit niedrigem Risiko eine vorhergesagte 1-Jahres-Überlebensrate von > 94 %, eine 1-Jahres-Überlebensrate mit mittlerem Risiko von 70 bis < 94 % und eine 1-Jahres-Überlebensrate mit hohem Risiko von < 70 %. Aktuelle Behandlungsrichtlinien integrieren eine Risikobewertung in den Algorithmus, um Behandlungspläne zu ändern.Stickoxid-WegStickoxid (NO) ist ein endogener Vasodilatator, der vom Lungenendothel produziert wird. Stickoxidsynthase (NOS) wandelt L-Arginin in NO um, das auf benachbarte glatte Muskelzellen einwirkt, wo es die Umwandlung von Guanosintriphosphat (GTP) in zyklisches Guanosinmonophosphat (cGMP) durch Guanylatcyclase katalysiert. Zyklisches GMP wiederum fördert die Entspannung der glatten Muskulatur und Vasodilatation, reguliert aber auch Zellproliferation, Apoptose und Entzündung. 126 , 127 Zyklisches GMP wird durch endogene Phosphodiesterase-5 (PDE-5) neutralisiert, wodurch seine Wirkung begrenzt wird. Patienten mit PAH haben eine mangelhafte NOS-Aktivität in den Lungengefäßen, was zu einer geringen NO-Produktion führt. 63 Günstige Ergebnisse bei der Behandlung von PAH wurden erzielt, indem der NO-cGMP-Weg mit Wirkstoffen angegriffen wurde, die PDE-5 hemmen (PDE-5-Inhibitoren) oder Guanylatcyclase direkt stimulieren (lösliche Guanylatcyclase-Stimulatoren).Phosphodiesterase-5-Inhibitoren begrenzen die Inaktivierung von cGMP und verstärken dadurch seine Wirkung auf die glatte Gefäßmuskulatur. Drei PDE-5-Hemmer, Sildenafil, Tadalafil und Vardenafil , verbessern nachweislich die pulmonale Hämodynamik, die Belastungsfähigkeit und die Symptome bei Patienten mit PAH, einschließlich solcher mit Bindegewebserkrankungen. 128–130 Außerdem hat sich gezeigt, dass Tadalafil die Zeit bis zur klinischen Verschlechterung verlängert. 129 Diese Mittel können eine schwere Hypotonie auslösen, wenn sie zusammen mit Nitroglycerin verwendet werden, und daher ist die Verwendung von Nitroglycerin mit PDE-5-Hemmern kontraindiziert. Die Sicherheit bei Schwangeren wurde nicht nachgewiesen; in Tierversuchen wu
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.
10 Mär 2023 12:03 #1734
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
danny antwortete auf PH und Therapien, Management bei Corona
Risikobewertung und Behandlung von PAH der WHO-Gruppe 1Die Behandlungsmöglichkeiten der pulmonalen arteriellen Hypertonie haben sich in den letzten Jahren rasant weiterentwickelt. Vor 1995 umfassten die Behandlungsoptionen für Patienten mit PAH Diuretika, Antikoagulation, Digoxin, Kalziumkanalblocker und Sauerstoff. Patienten mit ungehindertem Krankheitsverlauf könnten letztendlich eine Lungentransplantation benötigen. 1995 war Epoprostenol die erste zielgerichtete Therapie, die von der United States Federal Drug Administration für die Behandlung von PAH zugelassen wurde. Seitdem wurden mehrere zusätzliche zielgerichtete Therapien zugelassen, die die Zahl der nicht-chirurgischen Optionen für Patienten erweitern. Gegenwärtig verfügbare Behandlungsmittel für PAH zielen auf pathophysiologische Mechanismen in Stickoxid-, Endothelin- oder Prostacyclin-Wegen ab. Da immer mehr medizinische Therapien zur Verfügung stehen, Es gibt also Hinweise darauf, dass die Kombination von Wirkstoffen und die gleichzeitige Ausrichtung auf mehrere Signalwege oft vorteilhafter sind als deren einzelne Anwendung. Die Ausweitung des Einsatzes von zielgerichteten PAH-Therapien der WHO-Gruppe 1 zur Behandlung in anderen WHO-Gruppen war nur begrenzt erfolgreich, so dass es an guten Optionen für die Behandlung dieser Patienten mangelt. Daher wird sich der Fokus auf verfügbare Behandlungen in dieser Übersicht hauptsächlich auf Optionen für PAH der WHO-Gruppe 1 und CTEPH der Gruppe 4 beziehen, wobei neuere Untersuchungen zur Ausweitung der Mittel zur Verwendung in anderen WHO-Gruppen erwähnt werden.Die Auswahl aus den verfügbaren medizinischen Therapien zur Ausarbeitung eines wirksamen PAH-Behandlungsplans beinhaltet einen fortlaufenden Prozess der Risikobewertung und Ergebnisüberwachung. PAH ist eine fortschreitende Krankheit, selbst im gegenwärtigen Zeitalter der zielgerichteten Therapie, und eine sorgfältige Risiko- und Ergebnisüberwachung ist ein wesentlicher Bestandteil des Behandlungsprozesses. Die Beobachtung, dass es eine starke Korrelation zwischen dem Überleben und bestimmten klinischen Merkmalen bei PAH gibt, wie z. B. 6-Minuten-Gehstrecke, Funktionsklasse, hämodynamische Maße und andere Parameter, hat zur Entwicklung mehrerer Instrumente zur Risikobewertung geführt.
119
Die Funktion des rechten Ventrikels ist weithin als Determinante des Outcomes bei PAH anerkannt. Nichtinvasive Messungen der rechtsventrikulären Struktur und Funktion durch Echokardiographie oder Herz-MRT sind bei der Risikostratifizierung hilfreich und wurden in die Behandlungsleitlinien aufgenommen.
120
,
121
Die jüngsten Leitlinien der European Society of Cardiology/European Respiratory Society umfassen den Bereich des rechten Vorhofs, die systolische Exkursion in der Trikuspidalringebene/den systolischen Pulmonalarteriendruck (TAPSE/sPAP) und das Vorhandensein eines Perikardergusses unter den Risikostratifizierungsvariablen.
3
Risikobewertungs-Tools integrieren mehrere klinische Parameter, um vorherzusagen, ob ein Patient ein niedriges, mittleres oder hohes Risiko für einen kurzfristigen Tod durch PAH hat, und leiten somit den Ansatz für das medizinische Behandlungsdesign. Zwei der am häufigsten verwendeten Tools zur Risikobewertung sind der ESC/ERS-Risikobewertungsalgorithmus
3
und der REVEAL-Risikorechner.
122
Der REVEAL-Risikorechner wurde aus Daten erstellt, die im amerikanischen REVEAL-PAH-Register erfasst wurden, bei dem es sich um ein 3-jähriges Längsschnittregister von 2967 PAH-Patienten der WHO-Gruppe 1 mit gesammelten Daten zu klinischen Merkmalen, Bewertung, Behandlung und Ergebnissen handelte.
123
Eine validierte Überarbeitung des REVEAL-Rechners (REVEAL 2.0) enthält eine gekürzte Version (REVEAL Lite 2), die die Nützlichkeit in ambulanten Einrichtungen verbessert, in denen eine regelmäßige Nachsorge erfolgt.
124
,
125
Eine Risikobewertung wird in der Regel bei jedem regelmäßigen Nachsorgebesuch durchgeführt, und der Behandlungsplan wird nach Bedarf angepasst, um einen niedrigen Risikostatus zu erreichen und aufrechtzuerhalten. Basierend auf der REVEAL 2.0-Risikostratifizierung haben Patienten in der Gruppe mit niedrigem Risiko eine vorhergesagte 1-Jahres-Überlebensrate von > 94 %, eine 1-Jahres-Überlebensrate mit mittlerem Risiko von 70 bis < 94 % und eine 1-Jahres-Überlebensrate mit hohem Risiko von < 70 %. Aktuelle Behandlungsrichtlinien integrieren eine Risikobewertung in den Algorithmus, um Behandlungspläne zu ändern.Stickoxid-WegStickoxid (NO) ist ein endogener Vasodilatator, der vom Lungenendothel produziert wird. Stickoxidsynthase (NOS) wandelt L-Arginin in NO um, das auf benachbarte glatte Muskelzellen einwirkt, wo es die Umwandlung von Guanosintriphosphat (GTP) in zyklisches Guanosinmonophosphat (cGMP) durch Guanylatcyclase katalysiert. Zyklisches GMP wiederum fördert die Entspannung der glatten Muskulatur und Vasodilatation, reguliert aber auch Zellproliferation, Apoptose und Entzündung.
126
,
127
Zyklisches GMP wird durch endogene Phosphodiesterase-5 (PDE-5) neutralisiert, wodurch seine Wirkung begrenzt wird. Patienten mit PAH haben eine mangelhafte NOS-Aktivität in den Lungengefäßen, was zu einer geringen NO-Produktion führt.
63
Günstige Ergebnisse bei der Behandlung von PAH wurden erzielt, indem der NO-cGMP-Weg mit Wirkstoffen angegriffen wurde, die PDE-5 hemmen (PDE-5-Inhibitoren) oder Guanylatcyclase direkt stimulieren (lösliche Guanylatcyclase-Stimulatoren).Phosphodiesterase-5-Inhibitoren begrenzen die Inaktivierung von cGMP und verstärken dadurch seine Wirkung auf die glatte Gefäßmuskulatur. Drei PDE-5-Hemmer, Sildenafil, Tadalafil und Vardenafil , verbessern nachweislich die pulmonale Hämodynamik, die Belastungsfähigkeit und die Symptome bei Patienten mit PAH, einschließlich solcher mit Bindegewebserkrankungen.
128–130
Außerdem hat sich gezeigt, dass Tadalafil die Zeit bis zur klinischen Verschlechterung verlängert.
129
Diese Mittel können eine schwere Hypotonie auslösen, wenn sie zusammen mit Nitroglycerin verwendet werden, und daher ist die Verwendung von Nitroglycerin mit PDE-5-Hemmern kontraindiziert. Die Sicherheit bei Schwangeren wurde nicht nachgewiesen; in Tierversuchen wurde jedoch keine fötale Schädigung festgestellt.Guanylatcyclase kann durch Stimulatoren für lösliche Guanylatcyclase (sGC) direkt zur Produktion von cGMP stimuliert werden. Riociguat ist bisher der einzige sGC-Stimulator, der für die Behandlung von pulmonaler Hypertonie zugelassen ist, und wurde von der US-amerikanischen Federal Drug Administration sowohl für PAH als auch für CTEPH zugelassen. Der Nutzen von Riociguat bei pulmonaler Hypertonie wurde in zwei randomisierten klinischen Studien aufgeklärt. In PATENT-1 verbesserte Riociguat die Hämodynamik, die Belastungsfähigkeit und die Zeit bis zur klinischen Verschlechterung bei Patienten mit PAH.
131
In CHEST-1 war Riociguat mit einer signifikanten Verbesserung der körperlichen Leistungsfähigkeit und des pulmonalen Gefäßwiderstands bei Patienten mit CTEPH verbunden.
132
Riociguatist derzeit das einzige Therapeutikum, das für die Behandlung von CTEPH (WHO-Gruppe 4 PH) zugelassen ist. Wie die PDE-5-Hemmer kann Riociguat bei Anwendung mit Nitroglycerin Hypotonie verursachen. Daher ist die gemeinsame Anwendung von Riociguat und Nitroglycerin kontraindiziert. Darüber hinaus ist Riociguat teratogen und sollte nicht bei schwangeren Menschen angewendet werden. Frauen im gebärfähigen Alter müssen sich während der Einnahme von Riociguat monatlich einem Schwangerschaftstest unterziehen und sorgfältige Verhütungsmaßnahmen befolgen .Endothelin-WegEndothelin-1 ist ein Vasokonstriktor und mitogener Mediator, der von Endothelzellen produziert wird und in erhöhten Konzentrationen im Plasma und Lungengewebe von Patienten mit PAH vorhanden ist.
65
,
133
Endothelin übt seine Wirkung aus, indem es an zwei G-Protein-gekoppelte Rezeptoren vom Typ A und B bindet, die sich auf der Oberfläche glatter Muskelzellen befinden. Der Rezeptor vom Typ A vermittelt Vasokonstriktion, Zellwachstum und Entzündung, während der Rezeptor vom B-Typ die Vasodilatation und Natriurese fördert und gleichzeitig die Zellproliferation und Entzündung begrenzt. Es gibt drei Verbindungen in dieser Gruppe, die zur Behandlung von PAH verwendet werden, einschließlich Bosentan, Ambrisentan und Macitentan.Bosentan blockiert sowohl Typ-A- als auch Typ-B-Rezeptoren, um die Belastungsfähigkeit, die Zeit bis zur klinischen Verschlechterung, die Hämodynamik und echokardiographische Anomalien bei PAH zu verbessern.
134
Die Anwendung von Bosentan war in klinischen Studien bei etwa 10 % der Patienten mit einem Anstieg der Lebertransaminasen verbunden, was dazu führte, dass eine monatliche Überwachung der Leberfunktion erforderlich war. Darüber hinaus wurde die Bosentan- Exposition mit Ödemen und Anämie in Verbindung gebracht. Das Medikament ist teratogen und sollte nicht bei schwangeren Menschen angewendet werden. Frauen im gebärfähigen Alter müssen sich monatlich einem Schwangerschaftstest unterziehen und eine sorgfältige Empfängnisverhütung praktizieren.Im Gegensatz zu Bosentan ist Ambrisentan ein selektiver Typ-A-Rezeptorantagonist, der in klinischen Studien mit Patienten mit idiopathischer PAH oder PAH im Zusammenhang mit einer Bindegewebserkrankung oder einer HIV-Infektion mit einer signifikanten Verbesserung der Symptome, der körperlichen Leistungsfähigkeit, der klinischen Verschlechterung und der Hämodynamik in Verbindung gebracht wurde.
135
,
136
Das Risiko einer Leberschädigung ist bei Ambrisentan minimal , und monatliche Leberfunktionstests sind nicht erforderlich. Die Anwendung von Ambrisentan bei Patienten mit chronischer Lebererkrankung sollte jedoch vermieden werden. Auch hier ist Ambrisentan teratogen und führt zu denselben Schwangerschafts- und Empfängnisverhütungsmaßnahmen wie Bosentan .Der dritte und neueste Endothelin-Rezeptor-Antagonist ist Macitentan . Der Nutzen von Macitentan bei Patienten mit PAH wurde in einer großen ereignisgesteuerten Studie nachgewiesen, in der Macitentan das Risiko einer Krankheitsprogression um 45 % und das Risiko eines Todes oder einer Krankenhauseinweisung aufgrund von PAH um 50 % senkte.
137
Eine beträchtliche Anzahl von Patienten in dieser Studie erhielt eine Hintergrundtherapie mit anderen Wirkstoffen und zeigte dennoch einen signifikanten Nutzen von Macitentan . Die wichtigsten Nebenwirkungen dieses Medikaments sind Anämie und Flüssigkeitsretention. Eine Überwachung auf Leberschäden ist nicht erforderlich, jedoch wie andere Endothelin-Rezeptor-Antagonisten, Macitentanist teratogen und sollte nicht bei schwangeren Frauen angewendet werden. Bei gebärfähigen Frauen sind eine Überwachung der Schwangerschaft und sorgfältige Verhütungsmaßnahmen erforderlich.Prostacyclin-WegProstaglandin I2, das aus Arachidonsäure in den Lungenendothelzellen produziert wird, vermittelt die Relaxation glatter Muskelzellen, indem es die Produktion von zyklischem Adenosinmonophosphat (cAMP) aus Adenosintriphosphat (ATP) in glatten Muskelzellen katalysiert. Zyklisches AMP vermittelt die Relaxation der glatten Muskulatur, Vasodilatation und antiproliferative Aktivitäten im pulmonalen Gefäßbett. Die Prostacyclin-Analoga sind potente Vasodilatatoren des Lungengefäßsystems, Inhibitoren der Thrombozytenaggregation und Inhibitoren der Zellproliferation, die ihre Wirkung durch Wechselwirkung mit Prostaglandinrezeptoren entfalten.
138
Die pulmonale endotheliale Prostacyclin-Synthase-Expression ist bei PAH verringert, was zu einer verringerten Produktion von Prostaglandin I2 aus Arachidonsäure und dadurch zu einer verringerten Produktion von zyklischem AMP der glatten Muskulatur führt.
64
Synthetische Prostacyclin-Analoga können den Mangel an endogener Prostaglandin-I2-Produktion bei PAH ausgleichen und sind als orale, inhalative und subkutan oder intravenös verabreichte krankheitsmodifizierende Therapien erhältlich.Epoprostenol war das erste Prostacyclin-Analogon, von dem gezeigt wurde, dass es bei der Verringerung der Symptome, der Verbesserung der körperlichen Leistungsfähigkeit und der Verbesserung der Hämodynamik bei idiopathischer PAH und PAH im Zusammenhang mit systemischer Sklerose wirksam ist.
139
,
140
Darüber hinaus wurde in einer randomisierten Studie mit idiopathischen PAH-Patienten festgestellt, dass die Behandlung mit Epoprostenol die Sterblichkeit senkt.
140
Der anhaltende Langzeitnutzen von Epoprostenol wurde bei idiopathischer PAH, PAH in Verbindung mit anderen Erkrankungen und inoperabler CTEPH nachgewiesen.
141–145
Epoprostenolist nur als kontinuierliche IV-Therapie verfügbar und hat eine kurze Halbwertszeit von 3–5 Minuten. Tachyphylaxie tritt bei kontinuierlicher Infusion auf, die eine Dosissteigerung im Laufe der Zeit erfordert. Die Infusion wird in der Regel gut vertragen, obwohl Nebenwirkungen wie Kieferschmerzen, Kopfschmerzen, Hitzewallungen, Übelkeit und Durchfall für kurze Zeit nach einer Dosissteigerung auftreten können. Es besteht ein ernsthaftes Risiko einer Rebound-Vasokonstriktion und sogar des Todes, wenn die Infusion unterbrochen wird.Treprostinil ist ein neueres Prostacyclin-Analogon, das in oraler, inhalierter und subkutan oder intravenös infundierter Form erhältlich ist. Treprostinil wurde erstmals unter Verwendung einer subkutanen Verabreichungsmethode bei einer großen Gruppe von Patienten mit idiopathischer PAH und PAH im Zusammenhang mit Bindegewebserkrankungen untersucht. Obwohl die Dosistitration durch Nebenwirkungen wie Schmerzen an der Infusionsstelle begrenzt war, zeigte die Studie eine Verbesserung der Belastungstoleranz und der Hämodynamik.
146
,
147
Die Anwendung von Treprostinil bei PAH wurde später auf eine intravenöse Anwendung ausgedehnt. Die infundierten Formen von Treprostinil sind ähnlich wie Epoprostenol mit Tachyphylaxie verbunden, die eine kontinuierliche Überwachung und Dosiseskalation erfordert. Die Halbwertszeit von Treprostinil beträgt 3–4 Stunden. Die Nebenwirkungen ähneln denen, die bei Epoprostenol festgestellt wurden , und beinhalten das zusätzliche Risiko von Schmerzen an der Infusionsstelle bei Patienten, die den subkutanen Behandlungsweg verwenden. Die inhalative Darreichungsform von Treprostinil ist eine wirksame Behandlungsoption für Patienten, die möglicherweise nicht für ein kontinuierliches Infusionssystem geeignet sind. In einer randomisierten klinischen Studie mit inhalativem Treprostinil , an der Patienten teilnahmen, die bereits eine Basistherapie mit Bosentan oder Sildenafil erhielten, wurden signifikante Verbesserungen der 6-Minuten-Gehstrecke, der NT-proBNP-Spiegel und der Lebensqualitätsindikatoren beobachtet .
148
Inhaliertes Treprostinil wird viermal täglich verabreicht und ist als solches nicht mit der Entwicklung einer Tachyphylaxie verbunden. Inhaliertes Treprostinil wird gut vertragen, mit den häufigsten Nebenwirkungen wie Halsschmerzen, Husten und Kopfschmerzen. Eine orale Darreichungsform von Treprostinil hat auch in einer randomisierten klinischen Studie an behandlungsnaiven PAH-Patienten gezeigt, dass sie die 6-Minuten-Gehstrecke verbessert.
149
Orales Treprostinil wird nach einem zwei- oder dreimal täglich verabreichten Schema mit allmählicher Dosissteigerung verabreicht, um eine optimale Wirkung zu erzielen und aufrechtzuerhalten. Verwendung von oralem Treprostinilkann durch Anorexie, Übelkeit und Durchfall kompliziert werden. Die orale Darreichungsform sollte nicht bei Patienten mit Leberfunktionsstörung der Child-Pugh-Klasse 3 angewendet werden.Iloprost ist ein Prostacyclin-Analogon, das in Europa in intravenösen und inhalativen Formulierungen erhältlich ist, in den Vereinigten Staaten jedoch nur in inhalierter Form. Die inhalative Formulierung wurde bei Patienten mit PAH und CTEPH mit Placebo verglichen und führte zu einer Verbesserung der Symptome, der körperlichen Leistungsfähigkeit und der Hämodynamik.
150
Bei einer kleinen Gruppe von Patienten mit entweder PAH oder CTEPH wurde festgestellt, dass die Vorteile von intravenösem Iloprost denen von Epoprostenol ähnlich sind.
151
Inhaliertes Iloprostwird mit einem speziell entwickelten Vernebler geliefert und muss 6- bis 9-mal täglich behandelt werden. Jede Dosis ist 30–90 Minuten lang wirksam und kann mit Husten, Hitzewallungen oder Kieferschmerzen einhergehen. Hypotonie kann auftreten, daher ist bei Patienten mit niedrigerem Ausgangsblutdruck Vorsicht geboten.Neben der exogenen Verabreichung von Prostacyclin-Analoga kann die ungünstige Wirkung des Prostacyclin-Synthase-Mangels bei PAH auch mit einer neueren zielgerichteten Therapieklasse, dem Prostacyclin-Rezeptor-Agonisten, überwunden werden. Prostanoidrezeptoren innerhalb der Pulmonalarterie umfassen IP-, EP3- und TP-Rezeptoren. Der IP-Rezeptor vermittelt die Vasodilatation und begrenzt die Proliferation, während die anderen die Vasokonstriktion und die Zellproliferation fördern.
152–154
Bis heute ist Selexipag der einzige im Handel erhältliche Prostacyclinrezeptoragonist, obwohl ein zweiter Wirkstoff, Ralinepag , derzeit bei Patienten mit PAH untersucht wird.Selexipag wird zu einer aktiven Form metabolisiert, die 37-mal stärker ist als die Ausgangssubstanz. In einer ereignisgesteuerten Langzeitstudie reduzierte Selexipag das Risiko eines kombinierten Morbiditäts- und Mortalitätsendpunkts um 40 %.
155
,
156
Eine ähnliche Risikominderung des zusammengesetzten Endpunkts von 41 % wurde in einer Untergruppe von Patienten mit PAH im Zusammenhang mit einer Bindegewebserkrankung festgestellt.
157
Zusammengesetzte Endpunktereignisse umfassten die Notwendigkeit einer atrialen Septostomie oder Lungentransplantation, den Beginn einer chronischen Sauerstoff- oder parenteralen Prostanoidtherapie, einen Krankenhausaufenthalt aus PAH-bedingten Gründen, einen anderen Indikator für das Fortschreiten der Erkrankung oder den Tod. Eine große Mehrheit (80 %) der Patienten erhielt eine Basistherapie mit einem Endothelinrezeptorblocker, PDE-Hemmer oder beidem. Unabhängig von der Hintergrundtherapie wurde ein konsistenter Behandlungseffekt festgestellt. Häufige Nebenwirkungen dieses speziellen Mittels sind Kopfschmerzen, Hitzewallungen, Übelkeit, Durchfall, Myalgie und Arthralgie.KombinationstherapieWährend die verschiedenen therapeutischen Mittel, die zur Behandlung von PAH verwendet werden, an sich wirksam sind, haben sich Daten angesammelt, die darauf hindeuten, dass Kombinationen von Mitteln aus verschiedenen Klassen das Fortschreiten der Krankheit sogar noch wirksamer einschränken können. In den letzten Jahren wurden zahlreiche klinische Studien durchgeführt, die den Nutzen der Kombinationstherapie untersuchten und unterschiedliche Erfolgsgrade zeigten.
148
,
158
,
159
Eine der ausschlaggebenderen Studien zur Unterstützung der Kombinationstherapie war die Ambrisentan and Tadalafil in Patients with Pulmonary Hypertension (AMBITION)-Studie, in der die Kombinationstherapie mit Ambrisentan und Tadalafil im Voraus mit jedem Wirkstoff als Monotherapie verglichen wurde.
160
Die Kombination vonAmbrisentan und Tadalafil führten zu einer 50 %-igen Reduktion der kombinierten Morbidität/Mortalitätsereignisse im Vergleich zu einer alleinigen Anwendung der beiden Wirkstoffe. In einer retrospektiven Analyse von 106 Patienten verbesserte die Upfront-Kombination von Ambrisentan und Tadalafil den REVEAL 2.0-Risiko-Score, senkte den PVR und bewahrte das Schlagvolumen nach zweijähriger Behandlung, obwohl 57 % der Patienten in den mittleren oder hohen REVEAL 2.0-Risikokategorien blieben.
161
In der TRITON-Studie (The Efficacy and Safety of Initial Triple Versus Initial Dual Oral Combination Therapy in Patients With Newly Diagnosed Pulmonary Arterial Hypertension) wurden Patienten randomisiert, um eine anfängliche Kombinationstherapie mit Macitentan, Tadalafil und Placebo zu erhaltenMacitentan, Tadalafil und Selexipag .
162
In Woche 26 zeigten Patienten, die entweder eine Zweifach- oder Dreifachkombinationstherapie erhielten, eine signifikante Verbesserung des primären Endpunkts, PVR. Es gab jedoch keinen signifikanten Unterschied in der PVR zwischen den beiden Gruppen. Ebenso gab es eine Verbesserung bei den sekundären Endpunkten, 6-Minuten-Gehstrecke und NT-proBNP-Spiegel, aber auch hier kein signifikanter Unterschied zwischen den Gruppen. Die explorative Analyse deutete auf ein geringeres Risiko einer Krankheitsprogression in der Dreifach-Kombinationstherapie-Gruppe hin. Untersuchungen zu neueren Behandlungen wie Riociguat, Macitentan und Selexipag, haben eine beträchtliche Anzahl von Patienten mit Hintergrundtherapien eingeschlossen. Trotzdem haben diese Studien zusätzliche Verbesserungen der Symptome, der körperlichen Leistungsfähigkeit und der klinischen Verschlechterung gezeigt.
131
,
137
,
156
Signifikante Verbesserungen bei PVR, mPAP, Herzindex, rechtsatrialem Druck und gemischter venöser Sauerstoffsättigung wurden bei Kombinationstherapien berichtet.
163
Vorteile wie diese haben zur Aufnahme von Kombinationstherapieempfehlungen in aktuelle Behandlungsleitlinien geführt.
3
,
164
Chronische thromboembolische pulmonale HypertonieWährend sich ein Großteil der wissenschaftlichen Bemühungen in den letzten Jahren darauf konzentrierte, die Pathobiologie hinter PAH zu verstehen und wirksame krankheitsmodifizierende Behandlungen zu entwickeln, wurden auch Fortschritte beim Verständnis und der Behandlung von CTEPH erzielt. Die Pathogenese von CTEPH ist nicht genau definiert; es ist jedoch gekennzeichnet durch die Akkumulation von chronisch organisierten Thromben in den Lungenarterien in Verbindung mit Gefäßumbau, der eine ineffektive Fibrinolyse, endotheliale Dysfunktion und abnormale Angiogenese beinhalten kann, wie zuvor in diesem Review diskutiert.
165
CTEPH wird oft als Folge einer persistierenden Thromboembolie nach einer akuten Lungenembolie angesehen, jedoch liegen die Raten akuter Lungenembolien vor der Diagnose einer CTEPH je nach geografischer Lage zwischen 15 % und 75 %.
166
,
167
Eine aktuelle multizentrische beobachtende Kohortenstudie aus Deutschland (FOCUS-Studie) überwachte 1017 Patienten nach akuter Lungenembolie über 24 Monate und berichtete eine kumulative 2-Jahres-CTEPH-Inzidenz von 2,3 %.
168
Es ist klar, dass pulmonale Hypertonie in Verbindung mit chronischer Thromboembolie eine Krankheit sein kann, die nicht nur eine thrombotische Flussobstruktion, sondern auch einen fortschreitenden Gefäßumbau, einen steigenden pulmonalen Gefäßwiderstand und Rechtsherzversagen umfasst.CTEPH wird durch Rechtsherzkatheterisierung und Nachweis arterieller Okklusionen in Brustbildgebungsstudien nach mindestens 3 Monaten wirksamer Antikoagulation bestätigt. Die Diagnose kann schwierig sein, da den Symptomen nicht immer eine akute Lungenembolie vorausgeht. Die vorliegenden Symptome sind unspezifisch und können PAH oder andere kardiopulmonale Erkrankungen imitieren, wodurch die Erkennung von CTEPH häufig verzögert wird.
169
Die Diagnose hängt von der Rechtsherzkatheterisierung ab, die eine präkapilläre pulmonale Hypertonie und eine bildgebende Bestätigung bestätigt, die nicht übereinstimmende Perfusionsdefekte in der Ventilations-Perfusions-Bildgebung (VQ) oder charakteristische Befunde in der CT-Angiographie, MRT oder traditionellen Lungenangiographie umfassen können. VQ-Scanning ist das bevorzugte bildgebende Verfahren zur Diagnose von CTEPH mit einer berichteten Sensitivität von 96–97 % und einer Spezifität von 90–95 %.
170
Fortschritte in der CT-Angiographie haben die diagnostische Genauigkeit verbessert und sie in einer Studie mit VQ-Scanning vergleichbar gemacht, mit 100 % Sensitivität, 93,7 % Spezifität und 96,5 % Genauigkeit für VQ-Scanning im Vergleich zu 96,1 % Sensitivität, 95,2 % Spezifität und 95,6 % Genauigkeit bei CT-Angiographie .
171
Die chirurgische pulmonale Endarteriektomie (PEA) ist die empfohlene Behandlung der Wahl und kann für Patienten mit operabler Verschlusskrankheit heilbar sein. Fortschritte in der Operationstechnik ermöglichen in erfahrenen Zentren eine Endarteriektomie bis auf die Ebene subsegmentaler Äste. Perioperative Sterblichkeitsraten werden unter 2,5 % angegeben.
172–174
Die Langzeitüberlebensrate nach Endarteriektomie übertrifft in einem Bericht die nach nichtoperativer Behandlung mit einer 3-Jahres-Überlebensrate von 90 % nach Endarteriektomie und 70 % bei nichtoperativer Versorgung.
175
In ähnlicher Weise wurde eine 5-Jahres-Überlebensrate von 83 % bei Patienten mit proximaler Erkrankung berichtet, die sich einer Endarteriektomie unterzogen, im Vergleich zu 53 % bei Patienten, die eine Operation ablehnten.
176
Obwohl die Endarteriektomie in den meisten Fällen eine wirksame endgültige Behandlung für CTEPH darstellt, werden etwa 25 % der Patienten, die sich einer Operation unterziehen, eine anhaltende pulmonale Hypertonie haben.
177
Patienten, bei denen CTEPH diagnostiziert wurde, sollten in einem Zentrum mit Erfahrung in der pulmonalen Endarteriektomie untersucht werden, um festzustellen, ob sie für eine chirurgische Behandlung in Frage kommen.Für diejenigen Patienten, die möglicherweise keine Kandidaten für PEA sind, kann die Ballon-Lungenangioplastie (BPA) ein alternativer Behandlungsansatz sein. BPA wird als Option bei Patienten mit distaler thromboembolischer Erkrankung oder persistierender pulmonaler Hypertonie nach PEA angesehen. Es hat sich gezeigt, dass dieses Verfahren die Belastungskapazität, die Hämodynamik und die Funktion des rechten Ventrikels verbessert.
178–181
BPA wird in mehreren Sitzungen mit Dilatation einer begrenzten Anzahl von Läsionen pro Sitzung durchgeführt.
182
BPA kann als Zusatz bei Patienten mit anhaltender oder wiederkehrender pulmonaler Hypertonie nach PEA oder bei Patienten mit einseitiger Interoperabilität verwendet werden.
183
Obwohl das Verfahren in erfahrenen Händen effektiv ist, ist es nicht ohne signifikante potenzielle Komplikationen, einschließlich Gefäßverletzungen durch Drahtperforation oder Ballonüberdehnung, Lungenverletzung, Hämoptyse und Hypoxie. Die Entscheidung, BPA zu verfolgen, sollte Teil einer multidisziplinären Bewertung sein und nur in CTEPH-Zentren mit einem hohen Maß an BPA-Erfahrung durchgeführt werden.Ein erheblicher Anteil der CTEPH-Patienten kann als inoperabel angesehen werden oder sich gegen eine PEA entscheiden. Gezielte pulmonale Vasodilatatoren haben sich in dieser Population als vorteilhaft erwiesen. Signifikante Verbesserungen der 6-Minuten-Gehstrecke, des PVR, des NT-proBNP-Spiegels und der WHO-Funktionsklasse wurden in der CHEST-1-Studie beobachtet, in der Riociguat mit Placebo bei Patienten mit inoperabler CTEPH oder anhaltender pulmonaler Hypertonie nach PEA verglichen wurde.
132
Die Verlängerungsstudie CHEST-2 zeigte anhaltende 6-minütige Geh- und Funktionsklasse-Vorteile für bis zu 48 Monate.
184
In der Merit-1-Studie verbesserte Macitentan die 6-Minuten-Gehstrecke und den PVR in einer Phase-2-Untersuchung, in der Placebo mit 10 mg Macitentan verglichen wurdebei Patienten mit inoperabler CTEPH.
185
Die Sicherheit und Wirksamkeit von Macitentan 75 mg bei inoperabler CTEPH oder persistierender pulmonaler Hypertonie nach PEA wird derzeit in Phase 3 untersucht. Bei CTEPH-Patienten mit inoperabler Erkrankung oder persistierender pulmonaler Hypertonie nach Endarteriektomie hat sich auch gezeigt, dass subkutane Treprostini l die 6-Minuten-Gehstrecke verbessert.
186
Die Rolle der medizinischen Therapie in Verbindung mit PEA und BPA ist Gegenstand laufender Diskussionen und Untersuchungen. Es gibt einige Hinweise darauf, dass eine medizinische Behandlung als Überbrückung zu PEA die endgültige chirurgische Behandlung verzögern kann, ohne einen klinischen Nutzen zu bringen.
187
Im Gegensatz dazu können ausgewählte Patienten von einer Vorbehandlung mit gezielten PAH-Therapien profitieren, um die Hämodynamik vor der chirurgischen Endarteriektomie zu optimieren.
188
Behandlungsentwicklung in anderen WHO-GruppenMit dem Erfolg zielgerichteter Therapien bei Patienten der WHO-Gruppen 1 und 4 hat sich das Interesse entwickelt, ähnliche Lösungen für Patienten mit pulmonaler Hypertonie als Folge anderer Herz- und Lungenerkrankungen (WHO-Gruppen 2 und 3) zu finden. Wie bereits erwähnt, gibt es Merkmale des vaskulären Umbaus, die in allen WHO-Gruppen gemeinsam zu sein scheinen und wahrscheinlich aus einer pulmonalen Gefäßverletzung resultieren. Mehrere Studien, in denen gezielte PAH-Therapien bei Patienten der WHO-Gruppen 2 und 3 eingesetzt wurden, haben oft widersprüchliche und insgesamt enttäuschende Ergebnisse erbracht.Pulmonale Hypertonie aufgrund einer Linksherzerkrankung (WHO-Gruppe 2) ist die häufigste Form der pulmonalen Hypertonie und kann aus einer linksventrikulären systolischen oder diastolischen Dysfunktion, einer Herzklappenerkrankung oder angeborenen Störungen resultieren, die eine Einschränkung des linksventrikulären Ein- oder Ausflusses verursachen. Die Optimierung der Behandlung des Prozesses der primären Herzerkrankung bleibt der Eckpfeiler der Behandlung von Patienten der WHO-Gruppe 2. Obwohl mehrere Studien zielgerichtete PAH-Therapien eingesetzt haben, war der Erfolg bei der Suche nach einem geeigneten Ziel, um den Gefäßumbau bei dieser Patientengruppe sicher und effektiv umzukehren, aussichtslos. Eine kleine placebokontrollierte Studie mit Epoprostenol bei Patienten mit Herzinsuffizienz und reduzierter Ejektionsfraktion (HFrEF) zeigte eine Verbesserung der 6-Minuten-Gehstrecke ohne signifikante Nebenwirkungen.
189
Verbesserungen des Herzindex und des Kapillarkeildrucks, die in einer viel größeren internationalen Studie festgestellt wurden, in der HFrEF-Patienten randomisiert Epoprostenol oder Placebo erhielten, wurden durch eine erhöhte Sterblichkeit zunichte gemacht.
190
Studien mit Endothelin-Rezeptor-Antagonisten waren ebenfalls enttäuschend. Eine der bemerkenswertesten neueren Studien in dieser Hinsicht war die MELODY-1-Studie, in der Macitentan und Placebo bei Patienten mit kombinierter prä- und postkapillärer pulmonaler Hypertonie (CpcPH) verglichen wurden. Es gab keine signifikanten Verbesserungen der PVR-, PAWP- oder NT-proBNP-Spiegel mit Macitentan und Macitentan war mit einer signifikanten Flüssigkeitsretention verbunden.
191
Der Stickoxidweg wurde auch bei Patienten mit PH-LHD angegriffen. Die Wirkung des PDE-5-Hemmers Sildenafil hat einen positiveren Nutzen bei HFrEF als bei Patienten mit Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) gezeigt, wobei Studien Verbesserungen bei hämodynamischen Messungen, Sauerstoffverbrauch und körperlicher Leistungsfähigkeit zeigten.
192
Eine kleine Studie, in der Sildenafil und Placebo bei HFpEF verglichen wurden, konnte Verbesserungen des mPA-Drucks und der RV-Funktion bestätigen, jedoch nicht bei Atemnot oder Müdigkeit.
193
Eine weitere Studie, in der 60 mg Sildenafil dreimal täglich mit Placebo über 12 Wochen verglichen wurde, zeigte keine Verbesserung des primären Endpunkts, des mPA-Drucks.
194
Ähnlich in der RELAX-StudieSildenafil hatte keinen Einfluss auf die Aktivitätstoleranz oder Lebensqualität bei Patienten mit HFpEF.
195
Eine vergleichende Untersuchung von Riociguat und Placebo bei PH aufgrund von HFrEF zeigte eine Verbesserung des PVR und des Herzindex ohne Änderung des mPA-Drucks.
196
Eine zusätzliche Untersuchung von Modifikatoren des Stickoxid-Signalwegs für PH-LHD ist gerechtfertigt, um die Rolle dieser Mittel bei PH der WHO-Gruppe 2 zu klären.Wie im Fall von PH der WHO-Gruppe 2 gibt es nur begrenzte Daten zur Anwendung von PAH-Therapien zur Behandlung von PH der WHO-Gruppe 3, und die Ergebnisse in Studien, die sich mit dieser Population befassen, sind gemischt. Beispielsweise gibt es bei Patienten mit COPD Hinweise darauf, dass PAH-Therapien in einigen Fällen vorteilhaft, in anderen jedoch schädlich sein können. Verbesserungen der hämodynamischen Parameter und der Aktivitätstoleranz im Vergleich zum Ausgangswert wurden aus den ASPIRE- und COMPERA-Registern berichtet, wenn Patienten mit schwerer PH und COPD mit PDE-5-Inhibitoren behandelt wurden.
197
,
198
In einer großen randomisierten, kontrollierten, multizentrischen Studie mit Sildenafil wurden signifikante Verbesserungen der Hämodynamik, der körperlichen Leistungsfähigkeit und der Dyspnoe ohne Verschlechterung der Sauerstoffversorgung beobachtetim Vergleich zu Placebo bei Patienten mit schwerer PH und COPD.
199
Im Gegensatz dazu wurden in einer Studie mit Tadalafil bei Patienten mit fortgeschrittener COPD und leichter PH keine Verbesserungen der Aktivitätstoleranz oder der Lebensqualität beobachtet.
200
Die Anwendung von Bosentan in einer Patientengruppe mit leichter PH und schwerer COPD war mit einer Verschlechterung des Funktionsstatus und des Gasaustauschs verbunden.
201
Die Heterogenität von Patienten mit COPD und unterschiedlich starker Atemwegsobstruktion und Luftraumschädigung kann die widersprüchlichen Ergebnisse der bisherigen Studien erklären. Zukünftige Arbeiten in dieser Gruppe müssen sich möglicherweise auf bestimmte Subpopulationen mit heterogenen phänotypischen Manifestationen von COPD konzentrieren.Die Bemühungen, eine wirksame Behandlung für Patienten der WHO-Gruppe 3 mit interstitieller Lungenerkrankung (ILD) zu finden, haben ebenfalls zu gemischten Ergebnissen geführt. Eine klinische Studie zur Untersuchung der Anwendung von Riociguat bei PH in Verbindung mit idiopathischer Lungenfibrose wurde aufgrund einer zunehmenden klinischen Verschlechterung und des Mortalitätsrisikos abgebrochen.
202
Im Gegensatz zu diesen enttäuschenden Ergebnissen wurden in der INCREASE-Studie bemerkenswerte Vorteile beobachtet, in denen inhalatives Treprostinil mit Placebo bei Patienten mit PH-ILD verglichen wurde.
203
In dieser Studie wurden Patienten mit inhalativem Treprostinil behandelterlebten eine 31-Meter-Verbesserung bei 6-minütigem Gehen nach 16 Wochen, eine signifikante Verbesserung der NT-proBNP-Spiegel und weniger klinische Verschlechterungsereignisse im Vergleich zu Placebo. Die Anwendung von inhaliertem Treprostinil war auch mit einer unerwarteten Verbesserung der forcierten Vitalkapazität verbunden.Lungentransplantation und atriale SeptostomieObwohl Fortschritte in der medizinischen Therapie von PAH die Notwendigkeit einer Lungentransplantation in der PH-Patientenpopulation verringert haben, können diejenigen, bei denen die medizinische Therapie versagt, und diejenigen mit PH aufgrund anderer Lungenerkrankungen und Hypoxie Kandidaten für eine Lungentransplantation sein. Die bilaterale Lungentransplantation ist das Standardverfahren für PAH-Patienten. Zu den Indikationen für eine Transplantationsüberweisung gehören ein REVEAL-Risiko-Score >7 oder ein mittlerer oder hoher ESC/ERS-Risikostatus bei geeigneten PAH-Medikamenten, die Anwendung von parenteralen Prostanoiden, eine schnell fortschreitende Erkrankung und eine bestätigte oder vermutete pulmonale venöse Verschlusskrankheit oder pulmonale kapilläre Hämangiomatose und Anzeichen dafür Leber- oder Nierenfunktionsstörungen aufgrund von PAH.
204
Im Allgemeinen haben PAH-Patienten, die 1 Jahr nach der Transplantation überleben, eine mediane Überlebenszeit von 10 Jahren.
204
Eine kürzlich durchgeführte Studie berichtete über Überlebensraten von 93 % nach 3 Monaten, 90 % nach 1 Jahr, 87 % nach 3 Jahren und 87 % nach 5 Jahren.
205
Das Überleben hat sich mit Fortschritten im perioperativen Management verbessert, wie z. B. der Verwendung von ECMO anstelle eines kardiopulmonalen Bypasses.
206
Die Option einer Lungentransplantation sollte frühzeitig in Betracht gezogen werden, da diese Krankheitsentitäten schnell fortschreiten können und das Ansprechen auf aktuelle medizinische Therapien unvorhersehbar ist. Eine frühzeitige Überweisung ermöglicht eine vollständigere und sorgfältigere Bewertung vor der Transplantation und Zeit für Patienten, um die Anforderungen und Auswirkungen auf die Lunge zu berücksichtigen Transplantation auf sich und ihre Familien treffen wird. Eine atriale Septostomie ist als Überbrückung zur Lungentransplantation zu erwägen, wenn die medikamentöse Therapie versagt. Die Schaffung eines interatrialen Rechts-Links-Shunts kann dazu dienen, die rechte Herzkammer zu dekomprimieren und den Sauerstofftransport trotz Verringerung der Sauerstoffsättigung zu verbessern.
207
Die Herausforderung einer Covid-Infektion bei Patienten mit pulmonaler HypertonieIn den ersten Monaten der COVID-19-Pandemie tauchten Beobachtungsberichte auf, die darauf hindeuteten, dass Patienten mit PAH mit niedrigeren Krankenhausaufenthalts- und Sterblichkeitsraten möglicherweise besser abschneiden als die allgemeine Bevölkerung.
208–210
Daten einer Kohorte von 13 Patienten aus 32 US-amerikanischen PH-Zentren zeigten eine Hospitalisierungsrate von 53,8 % und eine Mortalität von 7,7 %.
208
Während die Hospitalisierungsrate in kleinen Kohorten aus Italien (100 % von 4 Patienten) und Spanien (70 % von 10 Patienten) höher war, lag die Sterblichkeit in beiden Fällen bei 0 %.
209
,
210
Mehrere Theorien wurden aufgestellt, um diese frühen Beobachtungen zu erklären, darunter endotheliale Schutzwirkungen gezielter PAH-Therapien, begrenzter Viruseintritt infolge einer geringeren Expression von Angiotensin-Converting-Enzym-2 (ACE-2) bei PAH und verringerte Reaktionsfähigkeit auf Veränderungen der Lungenperfusion in PAH.
211
Als sich jedoch die Erfahrungen mit COVID-19 bei Patienten mit PAH häuften, war es offensichtlich, dass die Ergebnisse weniger günstig waren als ursprünglich angenommen. Ende 2020 ging aus einer Umfrage in 47 PH-Zentren auf der ganzen Welt ein Bericht hervor, in dem vom 17. April 2020 bis zum 10. Mai 2020 insgesamt 70 Fälle von COVID-19-Infektionen bei PAH- und CTEPH-Patienten gemeldet wurden. In dieser Kohorte wurden 63 % ins Krankenhaus eingeliefert ( 46 % konventionelle Station, 17 % Intensivstation) und die Sterblichkeitsrate betrug 20 % bei Patienten mit PAH.
212
Eine weitere Umfrage wurde damals in den Vereinigten Staaten durchgeführt, bei der 50 Patienten mit PAH oder CTEPH und einer COVID-19-Infektion zwischen dem 17. April 2020 und dem 1. Mai 2020 von 58 PH-Zentrumsleitern identifiziert wurden.
213
In dieser Gruppe war die Inzidenz einer COVID-19-Infektion in der Patientenpopulation ähnlich wie in der Allgemeinbevölkerung, 30 % der Patienten wurden ins Krankenhaus eingeliefert, und 12 % überlebten nicht. Daten aus einer viel größeren Kohorte des French Pulmonary Hypertension Network mit 211 Patienten mit PH (123 PAH, 47 CTEPH, 41 andere PH) und einer COVID-19-Infektion vom 1. Februar 2020 bis 30. April 2021 lieferten zusätzliche Einblicke in die mit der Infektion verbundenen Ergebnisse in dieser Population.
214
Die Gesamtmortalität in dieser Kohorte betrug 24,6 %, die Krankenhausmortalität 41,3 %. Die Mehrheit der Patienten wurde ins Krankenhaus eingeliefert (59,7 %), wobei 32,2 % auf einer konventionellen Krankenhausstation behandelt wurden und 27,5 % eine Aufnahme auf der Intensivstation benötigten. Zu den Patientenmerkmalen, die mit einer höheren Sterblichkeit assoziiert sind, gehörten höheres Alter, männliches Geschlecht, Diabetes, Bluthochdruck, andere chronische Atemwegserkrankungen, chronische Herzerkrankungen und chronisches Nierenversagen. Die Anwendung der PAH-Therapie war bei Überlebenden und Nicht-Überlebenden ähnlich.Die endotheliale Dysfunktion ist von zentraler Bedeutung für die Entwicklung von PAH und kann durch Schäden, die durch die Virusinvasion von COVID-19 hervorgerufen werden, weiter verkompliziert werden. Eine Infektion mit SARS-CoV-2 verursacht diffuse alveoläre Schäden und perivaskuläre lymphozytäre Infiltration, aber auch spezifische Muster von Endothelverletzungen.
215
Zu den bemerkenswerten Befunden gehören Zellschwellungen, Kontaktverlust mit der Basalmembran und Unterbrechung der interzellulären Verbindungen. Es wird angenommen, dass das ACE-2-Protein der Endothelmembran ein Rezeptor für das COVID-19-Virus ist, das in Endothelzellen der Lungengefäße nachgewiesen wurde.
216
Zusätzlich zu den direkten Auswirkungen des Virus auf Endothelzellen zeigt die postmortale histologische Untersuchung Gefäßthrombosen, Mikroangiopathie und Verschlussbereiche im Alveolarkapillarbett.
217
,
218
Unter anderen Umständen gibt es Hinweise darauf, dass eine Störung der normalen vaskulären Endothelfunktion durch entzündliche und infektiöse Ereignisse zu einer Aktivierung thrombogener Wege, einem Versagen der normalen vasomotorischen Regulation zugunsten einer Vasokonstriktion, einer Beeinträchtigung der normalen endothelialen oxidativen Abwehr und einem Verlust der Barriere führen kann Integrität, was zu Kapillarlecks im mikrovaskulären Kompartiment führt.
219
Pathologische Auswirkungen einer COVID-19-Infektion auf die Biologie des pulmonalen Gefäßendothels könnten daher die endotheliale Dysfunktion bei Patienten mit PAH oder CTEPH verstärken.Patienten mit pulmonaler Hypertonie sollten ermutigt werden, eine COVID-19-Impfung zu erhalten. Sie sollten auch darauf hingewiesen werden, Maßnahmen zu ergreifen, die die Exposition gegenüber dem Virus im Alltag begrenzen können. Eine erfolgreiche Behandlung der pulmonalen Hypertonie hängt von der regelmäßigen Überwachung des klinischen Fortschritts des Patienten durch persönliche Besuche, Beurteilung der körperlichen Belastung und diagnostische Tests ab. Während des Höhepunkts der COVID-19-Pandemie begrenzten Telemedizinplattformen die Unterbrechungen der regelmäßigen Patientenbeurteilung bis zu einem gewissen Grad und ermöglichten es den Patienten, weiterhin wichtige Unterstützung von PH-Experten zu erhalten. Die Behandlung mit zielgerichteten PAH-Therapien sollte im Falle einer COVID-19-Infektion fortgesetzt werden. Während eine COVID-19-Infektion gemäß den aktuellen Behandlungsrichtlinien behandelt werden sollte,
211
Hochrisikopatienten, die ambulant behandelt werden können, aber anfällig für ungünstige Ergebnisse bei Infektionen sind, können von einer Behandlung mit neueren Virostatika wie Molnupiravir und Nirmatrelvir-Ritonavir profitieren. Es wurden keine signifikanten Wechselwirkungen zwischen Molnupiravir und derzeit verfügbaren PAH-Therapien berichtet. Nirmatrelvir-Ritonavir kann jedoch die Wirkung von Phosphodiesterase-5-Hemmern, Bosentan, Riociguat und Kalziumkanalblockern verändern. Die gleichzeitige Anwendung dieser PAH-Therapien und Nirmatrelvir-Ritonavir ist nicht ratsam. Bei Patienten mit schwerer Infektion kann eine Krankenhauseinweisung erforderlich sein, insbesondere wenn sie mit einer signifikanten Hypoxämie einhergeht. Während mit nasalen oder beheizten High-Flow-Kanülen eine ausreichende Oxygenierung erreicht werden kann, kann eine nicht-invasive Beatmung erforderlich sein, um ein hyperkapnisches Atemversagen auszugleichen. Intubation und Überdruckbeatmung können die rechtsventrikuläre Funktion weiter beeinträchtigen und sollten als letzter Ausweg zur Unterstützung des Gasaustauschs bei Patienten mit PAH und eingeschränkter rechtsventrikulärer Funktion in Betracht gezogen werden. Die Fortsetzung gezielter PAH-Therapien kann bei Krankenhauspatienten, die keine oralen Medikamente einnehmen können, eine Herausforderung darstellen und möglicherweise auf intravenöse oder inhalative Optionen umgestellt werden. Ein PAH-Experte kann wertvolle Managementhinweise geben und sollte konsultiert werden, wenn PAH-Patienten mit schweren COVID-19-Infektionen ins Krankenhaus eingeliefert werden. Expertenzentren sind oft in der Lage, Fernkonsultationen anzubieten oder die Verlegung von Patienten zu akzeptieren, wenn kein lokaler PAH-Experte verfügbar ist.Therapeutischer HorizontMehrere Therapeutika werden derzeit untersucht, die in naher Zukunft zusätzliche Optionen für die medizinische Behandlung von PAH bieten könnten. Ralinepag ist ein nicht-prostanoider, selektiver Prostacyclin (IP)-Rezeptoragonist, der derzeit in Phase 3 untersucht wird. In einer 22-wöchigen randomisierten, Placebo-kontrollierten Phase-2-Studie mit Patienten mit PAH unter einfacher oder doppelter oraler Hintergrundtherapie kam es bei Patienten, die Ralinepag erhielten, zu einer signifikanten Verringerung des PVR im Vergleich zu Placebo (–163,9 dyn-s-cm –5 mit Ralinepag vs 0,7 dyn-s-cm −5 mit Placebo, p = 0,02).
220
Verbesserungen der sekundären Endpunkte mit Ralinepag, einschließlich 6-Minuten-Gehstrecke, klinische Verschlechterung und NT-proBNP-Spiegel, waren statistisch nicht signifikant.Sotatercept ist ein erstes Fusionsprotein seiner Klasse, das zur Behandlung von PAH untersucht wird. Dieses neue Protein umfasst die extrazelluläre Domäne des menschlichen Aktivinrezeptors Typ IIA, fusioniert mit der Fc-Domäne von menschlichem IgG1. Sotatercept fungiert als Ligandenfalle für Mitglieder der Superfamilie des transformierenden Wachstumsfaktors Beta (TGF- und gleicht Wachstumsdifferenzierung und Hemmungswege aus. In der 24-wöchigen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Phase-2-Studie PULSAR wurden statistisch signifikante Verbesserungen des primären Endpunkts PVR bei Dosen von 0,3 und 0,7 mg im Vergleich zu Placebo festgestellt.
221
Reduktionen des PVR wurden bei Patienten mit einfacher, doppelter oder dreifacher Hintergrundtherapie, einschließlich infundierter Prostacycline, festgestellt. Darüber hinaus wurden Verbesserungen der 6-Minuten-Gehstrecke und der NT-proBNP-Spiegel beobachtet. Thrombozytopenie und erhöhte Hämoglobinspiegel waren bemerkenswerte Nebenwirkungen von besonderem Interesse in der PULSAR-Studie. Derzeit läuft eine Phase-3-Untersuchung.In vorläufigen Studien wurde gezeigt, dass Tyrosinkinase-Inhibitoren die hämodynamischen Parameter als Zusatzbehandlung für Patienten mit Erkrankungen verbessern, die auf Kombinationstherapien mit Stickoxid-, Endothelin- und/oder Prostacyclin-Pathway-Medikamenten nicht ansprechen. Es bestand beträchtliches Interesse an Imatinib , nachdem in einer placebokontrollierten Phase-2-Studie bei Patienten mit unzureichendem Ansprechen auf eine oder mehrere zielgerichtete PAH-Therapien signifikante Verbesserungen des PVR und des Herzzeitvolumens festgestellt wurden.
222
Imatinib hat eine hemmende Wirkung auf den Thrombozyten-Wachstumsfaktor (PDGF), von dem angenommen wird, dass er eine wichtige Rolle in der Pathobiologie von PAH spielt.
223
Weitere Studie zu Imatinib in der 24-wöchigen placebokontrollierten Imatinib -Studiebei pulmonaler arterieller Hypertonie bestätigte eine randomisierte Wirksamkeitsstudie (IMPRES) eine signifikante Verbesserung der 6-Minuten-Gehstrecke und PVR bei Patienten, die mit zwei oder mehr PAH-Therapien Imatinib erhielten .
224
Schwerwiegende Nebenwirkungen und Behandlungsabbrüche traten jedoch im Behandlungsarm erheblich häufiger auf, einschließlich zweier subduraler Hämatome bei Patienten, die Imatinib und Antikoagulation erhielten. Die Verlängerungsstudie wurde aufgrund häufiger schwerwiegender und unerwarteter unerwünschter Ereignisse, einschließlich sechs zusätzlicher subduraler Hämatome, vorzeitig abgebrochen.
225
Weitere Untersuchungen zu Imatinib werden derzeit durchgeführt, um die Vorteile einer neu formulierten orale
und gleicht Wachstumsdifferenzierung und Hemmungswege aus. In der 24-wöchigen, multizentrischen, randomisierten, doppelblinden, placebokontrollierten Phase-2-Studie PULSAR wurden statistisch signifikante Verbesserungen des primären Endpunkts PVR bei Dosen von 0,3 und 0,7 mg im Vergleich zu Placebo festgestellt.
221
Reduktionen des PVR wurden bei Patienten mit einfacher, doppelter oder dreifacher Hintergrundtherapie, einschließlich infundierter Prostacycline, festgestellt. Darüber hinaus wurden Verbesserungen der 6-Minuten-Gehstrecke und der NT-proBNP-Spiegel beobachtet. Thrombozytopenie und erhöhte Hämoglobinspiegel waren bemerkenswerte Nebenwirkungen von besonderem Interesse in der PULSAR-Studie. Derzeit läuft eine Phase-3-Untersuchung.In vorläufigen Studien wurde gezeigt, dass Tyrosinkinase-Inhibitoren die hämodynamischen Parameter als Zusatzbehandlung für Patienten mit Erkrankungen verbessern, die auf Kombinationstherapien mit Stickoxid-, Endothelin- und/oder Prostacyclin-Pathway-Medikamenten nicht ansprechen. Es bestand beträchtliches Interesse an Imatinib , nachdem in einer placebokontrollierten Phase-2-Studie bei Patienten mit unzureichendem Ansprechen auf eine oder mehrere zielgerichtete PAH-Therapien signifikante Verbesserungen des PVR und des Herzzeitvolumens festgestellt wurden.
222
Imatinib hat eine hemmende Wirkung auf den Thrombozyten-Wachstumsfaktor (PDGF), von dem angenommen wird, dass er eine wichtige Rolle in der Pathobiologie von PAH spielt.
223
Weitere Studie zu Imatinib in der 24-wöchigen placebokontrollierten Imatinib -Studiebei pulmonaler arterieller Hypertonie bestätigte eine randomisierte Wirksamkeitsstudie (IMPRES) eine signifikante Verbesserung der 6-Minuten-Gehstrecke und PVR bei Patienten, die mit zwei oder mehr PAH-Therapien Imatinib erhielten .
224
Schwerwiegende Nebenwirkungen und Behandlungsabbrüche traten jedoch im Behandlungsarm erheblich häufiger auf, einschließlich zweier subduraler Hämatome bei Patienten, die Imatinib und Antikoagulation erhielten. Die Verlängerungsstudie wurde aufgrund häufiger schwerwiegender und unerwarteter unerwünschter Ereignisse, einschließlich sechs zusätzlicher subduraler Hämatome, vorzeitig abgebrochen.
225
Weitere Untersuchungen zu Imatinib werden derzeit durchgeführt, um die Vorteile einer neu formulierten orale
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.