
- Beiträge: 1757
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Verträglichkeit, Sicherheit und Überleben bei Pat. mit schwerer PH /Veletri
Verträglichkeit, Sicherheit und Überleben bei Pat. mit schwerer PH /Veletri
15 Feb 2023 21:26 #1716
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Verträglichkeit, Sicherheit und Überleben bei Pat. mit schwerer PH /Veletri wurde erstellt von danny
respiratory-research.biomedcentral.com/a...tergrundEpoprostenol AS (Veletri ® ), eine thermostabile Epoprostenol-Formulierung, bietet im Vergleich zu früheren Epoprostenol-Formulierungen eine bessere Arzneimittelstabilität und eine verbesserte klinische Anwendung. Ziel dieser Studie ist es, die klinische Erfahrung in der Anwendung von Veletri ® zu erweitern , insbesondere in Bezug auf Verträglichkeit, Sicherheit und Überleben.MethodenPatienten mit pulmonaler arterieller Hypertonie (PAH) mit hohem Risiko trotz Vorbehandlung mit mindestens einer doppelten oralen Kombinationstherapie und mit klinischer Indikation für eine Behandlung mit Epoprostenol (Veletri ® ) wurden konsekutiv in diese prospektive, offene, nicht-interventionelle Beobachtungsstudie eingeschlossen. Klinische Daten wurden zu Studienbeginn, nach 3 und 6 Monaten bewertet. Unerwünschte Ereignisse (AEs) und schwerwiegende unerwünschte Ereignisse (SAEs) wurden dokumentiert. Das Überleben seit Beginn der Behandlung mit Veletri ® wurde beim letzten ausscheidenden Patienten beurteilt.ErgebnisseFünfzehn Patienten (60 ± 13,7 Jahre, WHO-Funktionsklasse III (n = 10) oder IV (n = 5), stark eingeschränkte rechtsventrikuläre Funktion, mittlerer pulmonalarterieller Druck 54,8 ± 8,9 mmHg, mittlerer pulmonaler Gefäßwiderstand 4,4 ± 0,7 (Median 3,8 ) Wood Units) wurden aufgenommen und mit einer mittleren Dosis von 7,9 ± 3,9 (Median 7,5) ng/kg/min behandelt. Elf Patienten beendeten die Studie (Behandlungsabbruch n = 1, Tod n = 3). Nach einer mittleren Nachbeobachtungszeit von 19,1 ± 13,5 (Median 18,0) Monaten starben sieben Patienten und drei wurden für eine Lungentransplantation gelistet. Sieben UE (Übelkeit n = 3, Durchfall n = 1, Flush n = 2, Kopfschmerzen n = 1) und drei SUE (Katheterinfektion n = 2, Katheterverschluss n = 1) standen im Zusammenhang mit Veletri ® . Die 1- und 2-Jahres-Überlebensraten betrugen 73,3 % bzw. 52,4 %.SchlussfolgerungenDie Studie zeigte, dass die Sicherheit und Verträglichkeit von Epoprostenol AS (Veletri ® ) mit früheren Prostacyclin-Formulierungen vergleichbar und für die meisten Patienten durchführbar war. Die maximal tolerierbare Dosierung war niedriger als die in der Literatur angegebenen Dosierungen. In zukünftigen Anwendungen/Studien sollte der Up-Titrationsprozess auf höhere Epoprostenol-Dosierungen beim Auftreten von Nebenwirkungen drängen, da das Erreichen einer hohen und wirksamen Dosierung entscheidend für den klinischen Nutzen der Patienten ist. Das Überleben war bei diesen weit verbreiteten schwer beeinträchtigten Patienten wie erwartet.Studienregistrierung Die Studie wurde im EUPAS-Register (EUPAS32492) registriert.HintergrundDie chronische pulmonale Hypertonie (PH) ist gekennzeichnet durch einen Anstieg des pulmonalvaskulären Widerstands und des pulmonalarteriellen Drucks, was zu Rechtsherzinsuffizienz und verschlechterter Prognose führt [
1
,
2
]. Eine Dysregulation der Prostacyclinsynthese mit resultierendem Mangel an endogenem Prostacyclin spielt eine wichtige Rolle in der Pathogenese der PH [
3
]. Prostacyclin hat gefäßerweiternde, antiproliferative, antiinflammatorische und antithrombotische Eigenschaften und ist daher eine wichtige Zielsubstanz in der PAH-spezifischen Therapie [
4
]. Epoprostenol war die erste spezifische Therapie, die für die Behandlung von PAH zugelassen wurde, nachdem positive Auswirkungen auf die Belastungsfähigkeit, wichtige hämodynamische Parameter, PAH-Symptome und das Überleben bei ansonsten behandlungsnaiven Patienten gezeigt wurden [ 2
,
5
]
. Epoprostenol ist bisher der einzige iv-Wirkstoff mit hohem Empfehlungsgrad für Patienten mit schwerer PAH, die in die Funktionsklasse der Weltgesundheitsorganisation (WHO-FC) III oder IV eingestuft sind [
1
,
6
].Epoprostenol, ein synthetisches Prostacyclin, ist bei Raumtemperatur chemisch instabil und hat eine kurze biologische Halbwertszeit von 3–5 min. Das Mittel kann einen schweren Rebound-PAH verursachen, wenn die Infusion abrupt unterbrochen wird [
1
]. Daher ist eine kontinuierliche intravenöse (iv) Infusion über einen zentralvenösen Verweilkatheter und eine externe Pumpe erforderlich. Folglich wird die Anwendung von Epoprostenol durch seine Handhabung behindert, was eine besondere Unbequemlichkeit für den Patienten bei der Aufbewahrung und Verabreichung des Medikaments darstellt [
7
].Epoprostenol, das Glycin und Mannitol (GM) enthält, wurde ursprünglich vor fast 20 Jahren in den USA für die Verwendung als langfristige kontinuierliche Infusion bei Patienten mit PAH zugelassen; diese Formulierung hat jedoch eine begrenzte Stabilität bei Raumtemperatur und erfordert die Verwendung von Kühlung oder häufige Medikationswechsel während der Verabreichung. Mit Veletri ® wurde eine verbesserte bioäquivalente Formulierung von intravenösem Epoprostenol entwickelt, die die Hilfsstoffe Arginin und Saccharose (Epoprostenol AS) enthält (Veletri ® , Caripul ® ). Es bietet eine bessere thermische Stabilität mit bis zu 72 h nach Rekonstitution je nach Konzentration, ist selbstkonservierend und lässt kein Wachstum von Mikroorganismen zu [
8
]. Außerdem frisch zubereitete Lösungen mit Veletri® ist gekühlt bis zu 8 Tage haltbar [
7
]. Während frühere Formulierungen von Epoprostenol GM wie Flolan ® ein spezielles Verdünnungsmittel zur Rekonstitution erforderten, kann Veletri ® entweder mit sterilem Wasser für Injektionszwecke oder mit 0,9 % Natriumchlorid-Injektion [
9
] rekonstituiert werden.Intravenöses Epoprostenol wird zur Behandlung von Erwachsenen mit schwerer pulmonaler arterieller Hypertonie (PAH) (Gruppe 1 der Weltgesundheitsorganisation) angewendet und ist seit Jahrzehnten weltweit im Einsatz. Allerdings wurde Veletri ® zu Beginn der Studie zu wenig in der klinischen Praxis eingesetzt und wird dies bis heute noch immer, obwohl verfügbare Daten einen Überlebensvorteil nahelegen, wenn es frühzeitig als Teil einer Kombinationstherapie angewendet wird [ 10
,
11
,
12
]
.Veletri ® ist die einzige in Deutschland erhältliche Epoprostenol-Formulierung mit sicherer 24-Stunden-Arzneimittelunterstützung. Da die intravenöse Behandlung immer noch komplex ist und Erfahrungen in der Verabreichung, Titration und Handhabung von iv Applikationssystemen selten sind, zielt diese Beobachtungsstudie darauf ab, die klinische Erfahrung und das Wissen in der Anwendung von Veletri ® bei Patienten mit schwerer PAH zu erweitern, insbesondere im Hinblick auf reale Lebensdaten zu Verträglichkeit, Sicherheit, klinischem Krankheitsverlauf und Überleben nach aktueller klinischer Praxis.MethodenPopulation und Design studierenDies war eine 6-monatige, offene, beobachtende, nicht-interventionelle prospektive Studie zur Bewertung der Anwendung, Sicherheit und Verträglichkeit von intravenösem Epoprostenol AS ( Veletri®). Fünfzehn Patienten mit invasiv diagnostizierter PAH durch Rechtsherzkatheter (mittlerer pulmonalarterieller Druck ≥ 25 mmHg in Ruhe, pulmonalarterieller Wedge-Druck ≤ 15 mmHg) der WHO-Funktionsklassen III und IV wurden zwischen 03/2018 und 08 konsekutiv in diese Beobachtungsstudie eingeschlossen /2020. Alle eingeschlossenen Patienten erhielten mindestens eine zweifache orale Kombinationstherapie mit PDE-5-Inhibitoren und ERA und benötigten eine Behandlungseskalation. Für die Studie in Frage kommende Patienten hatten entweder ein unbefriedigendes langfristiges klinisches Ansprechen oder gehörten trotz Zweifachkombinationsbehandlung immer noch zu einer Gruppe mit mittlerem oder hohem Risiko. Ausschlusskriterien waren eine bekannte Unverträglichkeit gegenüber Epoprostenol oder einem seiner sonstigen Bestandteile, Schwangerschaft oder Stillzeit. Darüber hinaus, Patienten, die innerhalb von 4 Wochen vor dem Screening an einer anderen klinischen Arzneimittelstudie teilnahmen, und/oder Patienten, die im Verlauf dieser Studie ein Prüfpräparat erhalten sollten, waren nicht teilnahmeberechtigt. Die Patienten gaben eine schriftliche Einverständniserklärung ab. Die Ethikkommission der Universität Heidelberg hatte keine Einwände gegen die Durchführung dieser Studie (S-699/2017). Die Studie entspricht der Deklaration von Helsinki in ihrer aktuellen Fassung. Die Studie wurde im EUPAS-Register (EUPAS32492) registriert.Die Patienten wurden zu Beginn der Behandlung sowie nach drei und sechs Monaten anhand routinemäßiger klinischer Untersuchungen beurteilt. Routineärztliche Untersuchungen, bestehend aus Anamnese, körperlicher Untersuchung, Elektrokardiogramm (EKG), Laboruntersuchungen [einschließlich N-terminalem prohirnigem natriuretischem Peptid (NT-proBNP)], 6-Minuten-Gehtest, Echokardiographie in Ruhe und Rechtsherzkatheter gemäß klinische Praxis. Um die Lebensqualität zu beurteilen, wurden die Teilnehmer gebeten, den SF-36-Fragebogen auszufüllen. Das Überleben und das transplantationsfreie Überleben wurden bei der 30-tägigen Nachuntersuchung nach dem letzten Studienuntersuchungsbesuch des Patienten bewertet.Eine Stichprobe von 15 Patienten sollte in die Studie aufgenommen werden, um Einblicke in die Anwendung, Sicherheit und Verträglichkeit der intravenösen Behandlung mit Epoprostenol (Veletri ® ) zu gewinnen und Daten zu sammeln.Die Daten wurden für jeden Patienten zu Beginn der intravenösen Behandlung mit Epoprostenol AS (Veletri ® ) und während des gesamten Beobachtungszeitraums aus den vorhandenen Krankenakten bei jedem klinischen Besuch auf dem jeweiligen Fallberichtsformular (CRF) gesammelt. Die Informationen wurden nach Verfügbarkeit und gemäß dem Besuchsplan für die klinische Praxis gesammelt. Die Daten wurden vom Studienpersonal in ein standardisiertes CRF eingegeben.MedikationsverwaltungUm einen systematischen Ansatz zu gewährleisten, wurden die behandelnden Ärzte über Empfehlungen für Behandlungsbeginn, Dosierung und Titration sowie über häufige Nebenwirkungen von intravenösem Epoprostenol informiert.Die Studienteilnehmer wurden zur Einleitung der Behandlung ins Krankenhaus eingeliefert, um eine kontinuierliche Titration von Veletri ® , eine angemessene Dosissteigerung und eine engmaschige Überwachung sicherzustellen. Abweichend von den offiziellen Dosierungsempfehlungen zur schnellen Einleitung einer chronischen Infusion von iv Epoprostenol (Veletri ® ) wurde die Medikation mit 2 ng/kg/min (oder niedriger bei Unverträglichkeit) begonnen und in Schritten von jeweils 1–2 ng/kg/min gesteigert 24 h, bis eine Toleranzgrenze des Medikaments festgestellt wurde.Die Dosis musste verringert werden, wenn dosislimitierende pharmakologische Wirkungen wie Übelkeit, Erbrechen, Hypotonie, Sepsis, Kopfschmerzen, Bauchschmerzen oder Atemnot auftraten. Während der Dosistitration wurden die Patienten durch Überwachung des Blutdrucks und der Herzfrequenz im Stehen und Liegen beobachtet, um die Verträglichkeit der Dosiserhöhung sicherzustellen.Im Falle von dosislimitierenden pharmakologischen Ereignissen wurde eine Verringerung der Infusionsgeschwindigkeit durchgeführt, falls dies als notwendig erachtet wurde; Die Dosis wurde schrittweise alle 15 Minuten oder länger in Schritten von 1 ng/kg/min verringert, bis die dosislimitierenden Wirkungen abklangen. Die Dosierung wurde aufrechterhalten, wenn die Nebenwirkungen nur leicht waren und spontan ausgesetzt wurden. Abruptes Absetzen von iv Epoprostenol (Veletri ® ) oder plötzliche starke Reduzierungen der Infusionsgeschwindigkeiten wurden strikt vermieden. Außer in lebensbedrohlichen Situationen (z. B. Bewusstlosigkeit, Kollaps usw.) durften die Infusionsraten von iv Epoprostenol (Veletri ® ) nur unter ärztlicher Anleitung angepasst werden.Veletri ® wurde, sobald es wie vorgeschrieben zubereitet war, durch kontinuierliche iv-Infusion über einen zentralen Venenkatheter und eine ambulante Infusionspumpe verabreicht.StatistikenDie Daten werden als Mittelwert ± Standardabweichung und 95 % Konfidenzintervall des Mittelwerts dargestellt. Für qualitative Daten, die n und % angeben, werden Häufigkeitstabellen bereitgestellt. Sicherheits- und Verträglichkeitsdaten von iv Epoprostenol wurden gesammelt und nach Schweregrad und Bezug zu Veletri ® nach Einschätzung des behandelnden Arztes aufgelistet. Der klinische Verlauf der Patienten wird anschaulich dargestellt. Es wurde eine Kaplan-Meier-Überlebensanalyse durchgeführt. Es gab keine Imputationsstrategie für fehlende Werte.
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.
15 Feb 2023 21:27 #1717
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Der erste Patient wurde im März 2018 aufgenommen. Der letzte Patient schloss die Studienbehandlung im März 2021 ab. Insgesamt 15 Patienten (60 ± 13,7 Jahre, 80 % weiblich, WHO-Funktionsklasse III–IV (Tabelle 1 ), stark eingeschränkte rechtsventrikuläre
Funktion
, mittlerer pulmonalarterieller Druck 54,8 ± 8,9 mmHg, mittlerer pulmonaler Gefäßwiderstand 4,4 ± 0,7 (Median 3,") Wood Units) wurden aufgenommen. Bei der Mehrheit war idiopathische pulmonale arterielle Hypertonie (IPAH) (66,6 %) oder PAH im Zusammenhang mit einer Bindegewebserkrankung (20 %, Tabelle
1
) diagnostiziert worden. Die mittlere Zeit von der Erstdiagnose bis zum Ausgangswert betrug 45,4 ± 23,7 Monate. Zu Beginn der Studie erhielten die meisten Patienten eine duale orale Kombinationsbehandlung (n = 14) mit PH-gerichteter Medikation in Übereinstimmung mit den Eignungskriterien (Tabelle
1
). Vier Patienten erhielten vor der Umstellung auf Veletri ® zusätzliche Prostazyklin-Analoga (inhalatives Iloprost n = 2, intravenöses Iloprost n = 1, subkutanes Treprostinil n = 1 ) . Patienten, die zuvor andere Prostacyclinanaloga wie Treprostinil oder Iloprost erhalten hatten, wurden auf Epoprostenol AS (Veletri ® ) umgestellt. Gründe für die Umstellung auf Veletri ® waren unzureichende Wirksamkeit bei den beiden Patienten mit inhalativem Iloprost und Probleme mit der Arzneimittelversorgung bei dem Patienten mit iv Iloprost. Der Patient, der zuvor Treprostinil erhielt, hatte wiederholt lokale Infektionen in der Nähe der Applikationsstelle und wurde daher auf Veletri ® umgestellt . Prostanoid-Behandlungen wurden über 3 Tage reduziert, um Veletri zu erhöhen® .Nach dem COMPERA-Ansatz befanden sich 14 Patienten in der mittleren Risikogruppe und einer in der Hochrisikogruppe. Sieben Patienten wiesen einen REVEAL-Risiko-Score ≥ 10 auf.Von 15 eingeschlossenen Patienten starben sieben Patienten und drei wurden während der Nachbeobachtungszeit für eine Lungentransplantation gelistet. Drei Patienten starben innerhalb des Studienzeitraums von 6 Monaten und vier Patienten starben während des Überlebens-Follow-up. Drei Patienten wurden für eine Lungentransplantation während der Überlebens-Follow-up-Periode aufgelistet.Elf Patienten beendeten die Studie (Behandlungsabbruch n = 1, Tod n = 3, Abb.
1
).Während der Studiendauer von 6 Monaten starben zwei Patienten einen Monat nach Behandlungsbeginn, einer 2½ Monate nach Behandlungsbeginn. Die Todesfälle waren auf multiples Organversagen aufgrund von Krebs im Endstadium (n = 2) und auf Rechtsherzversagen zurückzuführen. Abhängig von ihrem Allgemeinzustand wurden nicht alle Patienten bei jedem Studienbesuch allen geplanten Untersuchungen unterzogen. Ein Patient brach die Behandlung nach drei Monaten wegen Medikamentenunverträglichkeit und fehlendem Ansprechen auf die Behandlung mit gleichzeitiger Verschlechterung des PH ab. Dieser Patient starb etwa 3 Monate später. Ein Patient übersprang den Nachsorgetermin nach 3 Monaten und nahm am 6-Monats-Termin teil.Verwendung von Veletri®Veletri ® wurde im Krankenhaus über 7 ± 4 (1–12, Median 7) Tage titriert. Die Patienten wurden mit einer mittleren Maximaldosis von 7,9 ± 3,9 (3,4–20, Median 7,5) ng/kg/min behandelt, die meisten (12/15, 80 %) mit einer Dosis zwischen 5 und 10 ng/kg/min. Ein Patient, der eine deutlich höhere Dosierung von Veletri ® von 20 ng/kg/min erhielt, wurde zuvor mit Iloprost iv behandelt und konnte daher schneller und auf eine höhere verträgliche Dosis titriert werden (Abb.
2
). Weitere Dosierungen reichten von einer Höchstdosis von 3,4 ng/kg/min bis zu einer Höchstdosis von 10,4 ng/kg/min.Abb. 2
Wood Units) wurden aufgenommen. Bei der Mehrheit war idiopathische pulmonale arterielle Hypertonie (IPAH) (66,6 %) oder PAH im Zusammenhang mit einer Bindegewebserkrankung (20 %, Tabelle
1
) diagnostiziert worden. Die mittlere Zeit von der Erstdiagnose bis zum Ausgangswert betrug 45,4 ± 23,7 Monate. Zu Beginn der Studie erhielten die meisten Patienten eine duale orale Kombinationsbehandlung (n = 14) mit PH-gerichteter Medikation in Übereinstimmung mit den Eignungskriterien (Tabelle
1
). Vier Patienten erhielten vor der Umstellung auf Veletri ® zusätzliche Prostazyklin-Analoga (inhalatives Iloprost n = 2, intravenöses Iloprost n = 1, subkutanes Treprostinil n = 1 ) . Patienten, die zuvor andere Prostacyclinanaloga wie Treprostinil oder Iloprost erhalten hatten, wurden auf Epoprostenol AS (Veletri ® ) umgestellt. Gründe für die Umstellung auf Veletri ® waren unzureichende Wirksamkeit bei den beiden Patienten mit inhalativem Iloprost und Probleme mit der Arzneimittelversorgung bei dem Patienten mit iv Iloprost. Der Patient, der zuvor Treprostinil erhielt, hatte wiederholt lokale Infektionen in der Nähe der Applikationsstelle und wurde daher auf Veletri ® umgestellt . Prostanoid-Behandlungen wurden über 3 Tage reduziert, um Veletri zu erhöhen® .Nach dem COMPERA-Ansatz befanden sich 14 Patienten in der mittleren Risikogruppe und einer in der Hochrisikogruppe. Sieben Patienten wiesen einen REVEAL-Risiko-Score ≥ 10 auf.Von 15 eingeschlossenen Patienten starben sieben Patienten und drei wurden während der Nachbeobachtungszeit für eine Lungentransplantation gelistet. Drei Patienten starben innerhalb des Studienzeitraums von 6 Monaten und vier Patienten starben während des Überlebens-Follow-up. Drei Patienten wurden für eine Lungentransplantation während der Überlebens-Follow-up-Periode aufgelistet.Elf Patienten beendeten die Studie (Behandlungsabbruch n = 1, Tod n = 3, Abb.
1
).Während der Studiendauer von 6 Monaten starben zwei Patienten einen Monat nach Behandlungsbeginn, einer 2½ Monate nach Behandlungsbeginn. Die Todesfälle waren auf multiples Organversagen aufgrund von Krebs im Endstadium (n = 2) und auf Rechtsherzversagen zurückzuführen. Abhängig von ihrem Allgemeinzustand wurden nicht alle Patienten bei jedem Studienbesuch allen geplanten Untersuchungen unterzogen. Ein Patient brach die Behandlung nach drei Monaten wegen Medikamentenunverträglichkeit und fehlendem Ansprechen auf die Behandlung mit gleichzeitiger Verschlechterung des PH ab. Dieser Patient starb etwa 3 Monate später. Ein Patient übersprang den Nachsorgetermin nach 3 Monaten und nahm am 6-Monats-Termin teil.Verwendung von Veletri®Veletri ® wurde im Krankenhaus über 7 ± 4 (1–12, Median 7) Tage titriert. Die Patienten wurden mit einer mittleren Maximaldosis von 7,9 ± 3,9 (3,4–20, Median 7,5) ng/kg/min behandelt, die meisten (12/15, 80 %) mit einer Dosis zwischen 5 und 10 ng/kg/min. Ein Patient, der eine deutlich höhere Dosierung von Veletri ® von 20 ng/kg/min erhielt, wurde zuvor mit Iloprost iv behandelt und konnte daher schneller und auf eine höhere verträgliche Dosis titriert werden (Abb.
2
). Weitere Dosierungen reichten von einer Höchstdosis von 3,4 ng/kg/min bis zu einer Höchstdosis von 10,4 ng/kg/min.Abb. 2
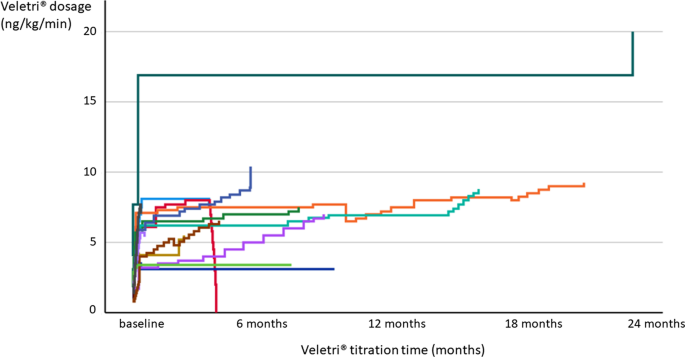 Dosistitration von Veletri ®
Die individuellen Erhaltungsdosen der Studienbehandlung variierten hauptsächlich zwischen 5 und 10 ng/kg/min.ÜberlebensanalyseNach einer mittleren Nachbeobachtungszeit von 19,1 ± 13,5 (Median 18,0) Monaten starben sieben Patienten und drei wurden für eine Lungentransplantation gelistet. Die transplantationsfreien 1- und 2-Jahres-Überlebensraten betrugen 73,3 % bzw. 52,4 % (Abb.
3
). Zusätzlich zu den drei Patienten, die innerhalb des Studienzeitraums von 6 Monaten starben, starben vier Patienten während der Nachbeobachtung. Todesursachen für Patienten, die während der Überlebenszeit verstarben, waren akutes Nierenversagen (n = 2), kardiopulmonales Versagen (n = 1) und Rechtsherzversagen (n = 1).Abb. 3
Dosistitration von Veletri ®
Die individuellen Erhaltungsdosen der Studienbehandlung variierten hauptsächlich zwischen 5 und 10 ng/kg/min.ÜberlebensanalyseNach einer mittleren Nachbeobachtungszeit von 19,1 ± 13,5 (Median 18,0) Monaten starben sieben Patienten und drei wurden für eine Lungentransplantation gelistet. Die transplantationsfreien 1- und 2-Jahres-Überlebensraten betrugen 73,3 % bzw. 52,4 % (Abb.
3
). Zusätzlich zu den drei Patienten, die innerhalb des Studienzeitraums von 6 Monaten starben, starben vier Patienten während der Nachbeobachtung. Todesursachen für Patienten, die während der Überlebenszeit verstarben, waren akutes Nierenversagen (n = 2), kardiopulmonales Versagen (n = 1) und Rechtsherzversagen (n = 1).Abb. 3
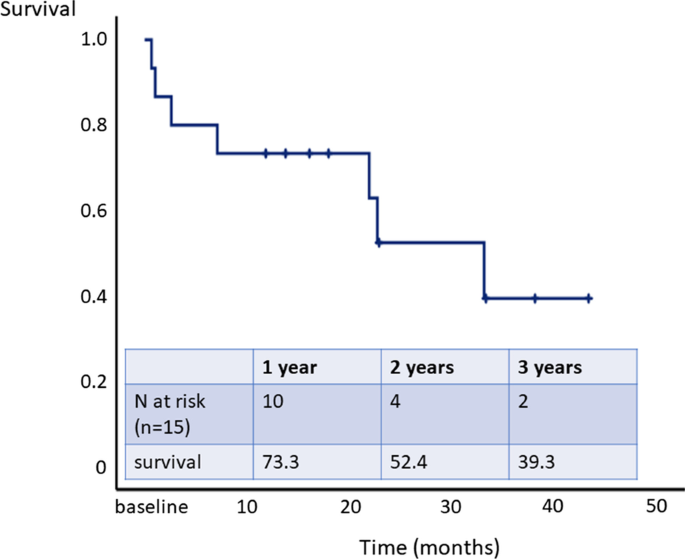 Überlebensanalyse
Die transplantationsfreien 1- und 2-Jahres-Überlebensraten betrugen 73,3 % bzw. 52,4 %.Sicherheit von Veletri®Bei 12 Patienten traten 16 schwerwiegende unerwünschte Ereignisse (SAE) auf. Drei SUE standen im Zusammenhang mit der Behandlung mit Veletri®: Katheterinfektion n = 2, Katheterverschluss n = 1 (Tabelle
2
). Die 10 SUE ohne Zusammenhang mit Veletri® umfassten zwei Todesfälle 30 Tage bzw. 77 Tage nach Behandlungsbeginn, die beide als nicht mit iv Epoprostenol zusammenhängend angesehen wurden (Abb. 1
)
. Ein weiterer Patient erlag 19 Tage nach Beginn der Behandlung mit iv Epoprostenol einer PAH-assoziierten Rechtsherzinsuffizienz.Tabelle 2 Sicherheitsanalyse
Alle drei Patientinnen, die innerhalb von 6 Monaten nach Studienbeginn starben, hatten eine Vorgeschichte von Krebs oder Verdacht auf Krebs (Verdacht auf einen Pseudotumor cerebri und eine mediastinale Raumforderung, Raumforderung im rechten unteren Lungenlappen, Brustkrebs in der Vorgeschichte). Darüber hinaus hatten alle drei Patienten eine arterielle Hypertonie, ein Patient eine koronare Herzkrankheit und ein Patient COPD II, GOLD B. Alle drei Patienten hatten zu Studienbeginn einen NT-proBNP > 12.000 pg/l. Sechs-Minuten-Fußweg war 20 und 201 m; Gehentfernung von einem Patienten wurde nicht erhalten. Diese Patienten wurden daher der WHO-Funktionsklasse IV zugeordnet. Zwei der drei Patienten starben an multiplem Organversagen, einer an Rechtsherzversagen.Von den anderen vier Patienten, die während der Langzeitnachsorge starben, hatten zwei eine begleitende systemische Sklerose, ein Patient eine Lungenembolie in der Vorgeschichte (jedoch kein CTEPH) und ein Patient litt an einer Leberzirrhose. Zwei Patienten starben an akutem Nierenversagen, zwei an Verdacht auf Rechtsherzversagen.Von dreizehn unerwünschten Ereignissen (AEs) standen 7 mit Veletri® in Zusammenhang : Übelkeit n = 3, Hitzewallungen n = 2, Durchfall n = 1 und Kopfschmerzen n = 1 (Tabelle
2
) traten innerhalb von 120,1 ± 87,3 Tagen nach Beginn der intravenösen Behandlung mit Epoprostenol auf. Einer der Patienten musste die Behandlung mit Epoprostenol aufgrund von Unverträglichkeit und Therapieresistenz nach der 3-monatigen Nachuntersuchung abbrechen. Die anderen 6 Epoprostenol-assoziierten UE waren ohne Krankenhausaufenthalt gut beherrschbar und konnten ohne Komplikationen oder Behandlungsunterbrechungen gebessert oder behoben werden. Bei zwei Patienten kam es zu Flush als dosislimitierendem UE, das bei reduzierter Dosierung zurückging.Klinischer Verlauf während der Veletri® - BehandlungDie Patienten wurden hinsichtlich ihrer körperlichen Belastbarkeit, Hämodynamik, echokardiographischen Parameter, Lungenfunktionstests und Lebensqualität beurteilt. Mittlere Änderungen der Parameter sind in Tabelle
3
angegeben .Tab. 3 Klinischer Verlauf während der Beobachtung
Acht Patienten konnten zu Studienbeginn einen 6-minütigen Gehtest durchführen. Alle Patienten hatten eine 6-minütige Gehstrecke (6MWD) unter 440 m. Zwei hatten eine Gehstrecke unter 165 m (ESC/ERS-Hochrisikozone). Zwei Patienten zeigten einen klinisch relevanten Anstieg von 6MWD > 35 m, ein Patient hatte einen Abfall von 47 m (Tabelle
3
). Die anderen Patienten blieben stabil. Kein Patient zeigte eine konstante Verbesserung der Gehstrecke in die ESC/ERS-Niedrigrisikokategorie.Zu Studienbeginn zeigten fünf Patienten ein erhöhtes NT-proBNP in der ESC/ERS-Zwischenrisikozone von 300–1400 ng/ml, sieben waren in der Hochrisikogruppe mit NT-proBNP von > 1400 ng/ml. Ein Patient verbesserte sich von mittlerem auf niedriges Risiko, einer von hohem auf mittleres Risiko. Ein Patient zeigte eine Verschlechterung von der mittleren zur hohen Risikokategorie für NT-proBNP.Von 12 Patienten mit mindestens zwei echokardiographischen Untersuchungen zeigten 9 eine Abnahme des systolischen pulmonalarteriellen Drucks im Bereich zwischen 5 und 50 mmHg.Bei acht Patienten lag der rechte Vorhofbereich in der mittleren Risikogruppe und bei sechs Patienten in der Hochrisikogruppe. Fünf Patienten zeigten im Verlauf der Studie eine Zunahme und fünf eine Abnahme des rechtsatrialen Bereichs. Ein Patient verbesserte die ESC/ERS-Risikogruppe für den rechten Vorhofbereich von mittlerem auf niedriges Risiko.Die Fläche des rechten Ventrikels nahm bei acht ab und bei vier Patienten zu. Die systolische Exkursion der Trikuspidalringebene nahm bei sechs zu (0,1–0,7 mm) und bei vier Patienten ab (– 0,2 bis – 0,8 mm).Rechtsventrikuläre Funktion und WHO-Funktionsklasse blieben bei allen Patienten im Studienverlauf stabil.Die Beurteilung der invasiven Hämodynamik durch Rechtsherzkatheterisierung konnte nur bei drei Patienten zu Studienbeginn und bei der Nachuntersuchung durchgeführt werden. Bei den anderen Patienten wurde vor Beginn der Studie eine kürzlich durchgeführte RHC durchgeführt. Alle drei Patienten zeigten einen Anstieg des pulmonalen Gefäßwiderstands.Eine stark eingeschränkte Lebensqualität [
13
] unter 50 Punkten zu Studienbeginn wurde bei elf Patienten für die physische Komponentenskala und bei neun Patienten für die mentale Komponentenskala festgestellt (Tabelle
3
).Zwei Patienten hatten eine Steigerung der körperlichen Lebensqualität um 30 Punkte im SF-36-Fragebogen. Die psychische Gesundheit verbesserte sich bei 6 Patienten um mehr als 10 Punkte. Von diesen Patienten zeigten drei eine anschließende Verschlechterung der psychischen Gesundheit. Bei zwei weiteren Patienten verschlechterte sich die psychische Gesundheit nach der Baseline.DiskussionDiese Studie gibt Einblick in die Sicherheit, Verträglichkeit und den klinischen Verlauf während der Behandlung mit Epoprostenol AS (Veletri ® ) unter realen Bedingungen. Die Behandlungsdosis von 7,9 ± 3,9 (3,4–20, Median 7,5) ng/kg/min war vergleichbar mit den in der Literatur berichteten Dosierungen einer medianen (Bereichs-)Dosis von 9,2 (8,0–15,0) ng/kg/min an Tag 28 [
14
]. Für die Langzeitbehandlung mit früheren Epoprostenol-Formulierungen (z. B. Flolan®) wurden jedoch deutlich höhere Dosierungen von bis zu 30–40 ng/kg/min berichtet [ 15
]
.Die höchste Dosierung wurde bei einem Patienten erreicht, der zuvor Iloprost iv erhalten hatte und auf Veletri ® umgestellt wurde , was höchstwahrscheinlich auf einen Gewöhnungseffekt des Medikaments zurückzuführen ist, der eine höhere Dosierung im Langzeitgebrauch erfordert. Die EPITOME-2-Studie deutet auf eine ähnliche kurzfristige Sicherheit und Wirksamkeit von Veletri ® bei PAH-Patienten hin, die stabil auf eine Flolan ® -Langzeittherapie eingestellt sind. Allerdings wurden deutlich höhere Dosen mit einem Mittelwert von 29,9 ± 15,1 ng/kg/min zu Studienbeginn und 30,2 ± 15,0 ng/kg/min nach drei Monaten verwendet [
7
]. In der kontrollierten 12-wöchigen Studie bei PAH wurde die Dosis von einer mittleren Anfangsdosis von 2,2 ng/kg/min erhöht. Während der ersten 7 Behandlungstage wurde die Dosis täglich auf eine mittlere Dosis von 4,1 ng/kg/min am 7. Behandlungstag erhöht. Am Ende von Woche 12 betrug die mittlere Dosis 11,2 ng/kg/min. Die mittlere schrittweise Erhöhung betrug 2–3 ng/kg/min alle 3 Wochen.Das Sicherheitsprofil in unserer Studie war mit Daten in der Literatur vergleichbar, wobei Übelkeit, Durchfall, Hitzewallungen und Kopfschmerzen zu den häufigsten unerwünschten Ereignissen im Zusammenhang mit dem Medikament gehörten [
2
].In Bezug auf die klinische Wirksamkeit gab es keine allgemeine Tendenz zur Verbesserung der gemessenen Parameter zur Beurteilung der körperlichen Leistungsfähigkeit (6MWD, Borg-Skala), der Lebensqualität (Kurzform-Gesundheitsbefragung 36; SF-36), der Hämodynamik, der rechten Herzgröße und Funktion sowie Lungenfunktion. Auch die WHO-FC blieb über den Untersuchungszeitraum stabil.Obwohl in dieser Studie keine klinisch relevante Verbesserung beobachtet wurde, blieben die meisten Patienten unter der Veletri® - Therapie stabil. In der Literatur wurde bereits über einen stabilen klinischen Verlauf unter Veletri ® -Behandlung berichtet, dies wurde jedoch bei Patienten beobachtet, die von Epoprostenol GM auf Epoprostenol AS umgestellt wurden [
7
,
15
,
16
]. Patienten, die von Epoprostenol GM auf Epoprostenol AS umgestellt wurden, berichteten von einem verbesserten Behandlungskomfort [
17
], während sich andere Parameter der Lebensqualität nicht veränderten. Obwohl in dieser Studie keine Änderung des Behandlungsschemas vorgenommen wurde, verbesserte sich die psychische Gesundheit bei zehn von fünfzehn Patienten.Bei Patienten mit neu begonnener Behandlung mit Epoprostenol umfassen die Behandlungseffekte eine Verbesserung der Hämodynamik [
5
,
18
,
19
], eine Verbesserung der körperlichen Belastbarkeit [
5
,
20
,
21
] und eine Verbesserung der Lebensqualität [
5
,
22
]. In unserer Kohorte konnten diese Effekte nicht beobachtet werden. Dies könnte auf die Schwere der Erkrankung zu Studienbeginn zurückzuführen sein, da fast die Hälfte der Studienkohorte im Verlauf der Studie und der Nachbeobachtung starb. Möglicherweise war die verabreichte Dosis von Veletri ® zu niedrig. Alle verstorbenen Patienten hatten jedoch Grunderkrankungen, die auf ein höheres Sterblichkeitsrisiko zurückzuführen sein könnten.Die Mehrheit der PH-bedingten UE (vier von sechs) wurde innerhalb von 3 Monaten nach Therapiebeginn beobachtet (Mittelwert: 120,2 ± 87,39 Tage, 95 %-KI: 28,6–211, und besserte sich im Laufe des Verlaufs, mit Ausnahme eines Todesfalls 19 Tage danach Veletri® - Initiation.Stärken und Grenzen der StudieDiese Studie wurde entwickelt, um Erkenntnisse über die klinische Praxis, Sicherheit, Verträglichkeit und den klinischen Verlauf während der Behandlung mit Veletri ® zu gewinnen . Ein beobachtendes Design ist vorzuziehen, wenn man darauf abzielt, Daten aus der realen Welt zu sammeln.Die Ergebnisse dieser Studie müssen unter Berücksichtigung ihrer potenziellen Einschränkungen aufgrund der geringen Anzahl von Teilnehmern und ihres nicht-interventionellen Designs betrachtet werden, das anfällig für Verluste bei der Nachverfolgung oder Abbrüche ist, die sich auf die Ergebnisse sowie die Signifikanz auswirken. Behandlungseffekte können aufgrund fehlender Werte von Patienten, die sich nicht bei jedem Besuch allen geplanten Untersuchungen unterzogen haben, möglicherweise nicht genau abgeschätzt werden. Darüber hinaus übersprang ein Patient die 3-monatige Nachbeobachtung, ein anderer Patient brach die Studie nach 3-monatiger Nachbeobachtung ab und drei Patienten starben während des 24-wöchigen Studienzeitraums aufgrund von Ursachen, die wahrscheinlich nicht mit iv Epoprostenol zusammenhängen. Insbesondere Änderungen der Endpunkte der Rechtsherzkatheterisierung konnten nicht zuverlässig bestimmt werden, da sich nur drei Patienten dieser Untersuchung zum 6-Monats-Follow-up unterzogen.Möglicherweise wurden Dosiserhöhungen zu niedrig durchgeführt. Während des stationären Aufenthaltes wurde Epoprostenol mit 2 ng/kg/min begonnen und alle 24 h um 1–2 ng/kg/min gesteigert, bis nach 5 Tagen eine Dosis von 10 ng/kg/min erreicht war (in Krankenhaus). Danach sollte die Dosis alle 1–2 Wochen um 1–2 ng/kg/min erhöht werden, um nach 6–8 Wochen 16 ng/kg/min zu erreichen. Bei den meisten Patienten haben wir diese Dosierung nicht erreicht. Möglicherweise haben wir nach dem stationären Therapiebeginn die Dosierung nicht ausreichend erhöht.Ziel der Studie war es, die klinische Erfahrung und das Wissen zu erweitern. Die Dosistitration ist ein wichtiger Teil, der in zukünftigen Anwendungen verbessert werden könnte und den wir vom PAH-Zentrum im Hôpital Bicêtre in Frankreich gelernt haben.Eine weitere Einschränkung dieser Studie ist der monozentrische Charakter dieser Studie. Daher kann ein Selektionsbias bei der Patientenrekrutierung nicht ausgeschlossen werden. Zudem war diese Studie als einarmige Open-Label-Studie geplant, weshalb eine Kontrollgruppe zum direkten Vergleich fehlt.Darüber hinaus kann die Verwendung von Fragebögen zu einer Verzerrung geführt haben, da Personen je nach Allgemeinzustand möglicherweise nicht gleichermaßen in der Lage sind, einen Fragebogen auszufüllen, was sich wiederum auf die aufgezeichneten Ergebnisse auswirkt.Im Allgemeinen war der Beobachtungszeitraum dieser Studie nicht lang genug, um die langfristigen Behandlungseffekte und das Überleben bei PAH-Patienten zuverlässig zu bestimmen. Weitere Studien mit einer größeren Teilnehmerzahl und einem verlängerten Überlebenszeitraum sind daher notwendig, um Titration, Dosierung und Dosiseskalation zu optimieren und die Wirksamkeit in Bezug auf das (transplantationsfreie) Überleben bei Patienten mit PAH zu bestimmen, um letztendlich eine Standardisierung zu leiten der Langzeittherapie.SchlussfolgerungenDie Studie zeigt, dass die Behandlung mit Epoprostenol (Veletri ® ) keine neuen Sicherheits- oder Verträglichkeitsaspekte ergab und bei den meisten Patienten durchführbar war. Die Überlebensrate war bei diesen schwer beeinträchtigten Patienten wie erwartet. In zukünftigen Anwendungen/Studien sollte der Up-Titrationsprozess auf höhere Epoprostenol-Dosierungen beim Auftreten von Nebenwirkungen drängen, da das Erreichen einer hohen und wirksamen Dosierung entscheidend für den klinischen Nutzen der Patienten ist.
Überlebensanalyse
Die transplantationsfreien 1- und 2-Jahres-Überlebensraten betrugen 73,3 % bzw. 52,4 %.Sicherheit von Veletri®Bei 12 Patienten traten 16 schwerwiegende unerwünschte Ereignisse (SAE) auf. Drei SUE standen im Zusammenhang mit der Behandlung mit Veletri®: Katheterinfektion n = 2, Katheterverschluss n = 1 (Tabelle
2
). Die 10 SUE ohne Zusammenhang mit Veletri® umfassten zwei Todesfälle 30 Tage bzw. 77 Tage nach Behandlungsbeginn, die beide als nicht mit iv Epoprostenol zusammenhängend angesehen wurden (Abb. 1
)
. Ein weiterer Patient erlag 19 Tage nach Beginn der Behandlung mit iv Epoprostenol einer PAH-assoziierten Rechtsherzinsuffizienz.Tabelle 2 Sicherheitsanalyse
Alle drei Patientinnen, die innerhalb von 6 Monaten nach Studienbeginn starben, hatten eine Vorgeschichte von Krebs oder Verdacht auf Krebs (Verdacht auf einen Pseudotumor cerebri und eine mediastinale Raumforderung, Raumforderung im rechten unteren Lungenlappen, Brustkrebs in der Vorgeschichte). Darüber hinaus hatten alle drei Patienten eine arterielle Hypertonie, ein Patient eine koronare Herzkrankheit und ein Patient COPD II, GOLD B. Alle drei Patienten hatten zu Studienbeginn einen NT-proBNP > 12.000 pg/l. Sechs-Minuten-Fußweg war 20 und 201 m; Gehentfernung von einem Patienten wurde nicht erhalten. Diese Patienten wurden daher der WHO-Funktionsklasse IV zugeordnet. Zwei der drei Patienten starben an multiplem Organversagen, einer an Rechtsherzversagen.Von den anderen vier Patienten, die während der Langzeitnachsorge starben, hatten zwei eine begleitende systemische Sklerose, ein Patient eine Lungenembolie in der Vorgeschichte (jedoch kein CTEPH) und ein Patient litt an einer Leberzirrhose. Zwei Patienten starben an akutem Nierenversagen, zwei an Verdacht auf Rechtsherzversagen.Von dreizehn unerwünschten Ereignissen (AEs) standen 7 mit Veletri® in Zusammenhang : Übelkeit n = 3, Hitzewallungen n = 2, Durchfall n = 1 und Kopfschmerzen n = 1 (Tabelle
2
) traten innerhalb von 120,1 ± 87,3 Tagen nach Beginn der intravenösen Behandlung mit Epoprostenol auf. Einer der Patienten musste die Behandlung mit Epoprostenol aufgrund von Unverträglichkeit und Therapieresistenz nach der 3-monatigen Nachuntersuchung abbrechen. Die anderen 6 Epoprostenol-assoziierten UE waren ohne Krankenhausaufenthalt gut beherrschbar und konnten ohne Komplikationen oder Behandlungsunterbrechungen gebessert oder behoben werden. Bei zwei Patienten kam es zu Flush als dosislimitierendem UE, das bei reduzierter Dosierung zurückging.Klinischer Verlauf während der Veletri® - BehandlungDie Patienten wurden hinsichtlich ihrer körperlichen Belastbarkeit, Hämodynamik, echokardiographischen Parameter, Lungenfunktionstests und Lebensqualität beurteilt. Mittlere Änderungen der Parameter sind in Tabelle
3
angegeben .Tab. 3 Klinischer Verlauf während der Beobachtung
Acht Patienten konnten zu Studienbeginn einen 6-minütigen Gehtest durchführen. Alle Patienten hatten eine 6-minütige Gehstrecke (6MWD) unter 440 m. Zwei hatten eine Gehstrecke unter 165 m (ESC/ERS-Hochrisikozone). Zwei Patienten zeigten einen klinisch relevanten Anstieg von 6MWD > 35 m, ein Patient hatte einen Abfall von 47 m (Tabelle
3
). Die anderen Patienten blieben stabil. Kein Patient zeigte eine konstante Verbesserung der Gehstrecke in die ESC/ERS-Niedrigrisikokategorie.Zu Studienbeginn zeigten fünf Patienten ein erhöhtes NT-proBNP in der ESC/ERS-Zwischenrisikozone von 300–1400 ng/ml, sieben waren in der Hochrisikogruppe mit NT-proBNP von > 1400 ng/ml. Ein Patient verbesserte sich von mittlerem auf niedriges Risiko, einer von hohem auf mittleres Risiko. Ein Patient zeigte eine Verschlechterung von der mittleren zur hohen Risikokategorie für NT-proBNP.Von 12 Patienten mit mindestens zwei echokardiographischen Untersuchungen zeigten 9 eine Abnahme des systolischen pulmonalarteriellen Drucks im Bereich zwischen 5 und 50 mmHg.Bei acht Patienten lag der rechte Vorhofbereich in der mittleren Risikogruppe und bei sechs Patienten in der Hochrisikogruppe. Fünf Patienten zeigten im Verlauf der Studie eine Zunahme und fünf eine Abnahme des rechtsatrialen Bereichs. Ein Patient verbesserte die ESC/ERS-Risikogruppe für den rechten Vorhofbereich von mittlerem auf niedriges Risiko.Die Fläche des rechten Ventrikels nahm bei acht ab und bei vier Patienten zu. Die systolische Exkursion der Trikuspidalringebene nahm bei sechs zu (0,1–0,7 mm) und bei vier Patienten ab (– 0,2 bis – 0,8 mm).Rechtsventrikuläre Funktion und WHO-Funktionsklasse blieben bei allen Patienten im Studienverlauf stabil.Die Beurteilung der invasiven Hämodynamik durch Rechtsherzkatheterisierung konnte nur bei drei Patienten zu Studienbeginn und bei der Nachuntersuchung durchgeführt werden. Bei den anderen Patienten wurde vor Beginn der Studie eine kürzlich durchgeführte RHC durchgeführt. Alle drei Patienten zeigten einen Anstieg des pulmonalen Gefäßwiderstands.Eine stark eingeschränkte Lebensqualität [
13
] unter 50 Punkten zu Studienbeginn wurde bei elf Patienten für die physische Komponentenskala und bei neun Patienten für die mentale Komponentenskala festgestellt (Tabelle
3
).Zwei Patienten hatten eine Steigerung der körperlichen Lebensqualität um 30 Punkte im SF-36-Fragebogen. Die psychische Gesundheit verbesserte sich bei 6 Patienten um mehr als 10 Punkte. Von diesen Patienten zeigten drei eine anschließende Verschlechterung der psychischen Gesundheit. Bei zwei weiteren Patienten verschlechterte sich die psychische Gesundheit nach der Baseline.DiskussionDiese Studie gibt Einblick in die Sicherheit, Verträglichkeit und den klinischen Verlauf während der Behandlung mit Epoprostenol AS (Veletri ® ) unter realen Bedingungen. Die Behandlungsdosis von 7,9 ± 3,9 (3,4–20, Median 7,5) ng/kg/min war vergleichbar mit den in der Literatur berichteten Dosierungen einer medianen (Bereichs-)Dosis von 9,2 (8,0–15,0) ng/kg/min an Tag 28 [
14
]. Für die Langzeitbehandlung mit früheren Epoprostenol-Formulierungen (z. B. Flolan®) wurden jedoch deutlich höhere Dosierungen von bis zu 30–40 ng/kg/min berichtet [ 15
]
.Die höchste Dosierung wurde bei einem Patienten erreicht, der zuvor Iloprost iv erhalten hatte und auf Veletri ® umgestellt wurde , was höchstwahrscheinlich auf einen Gewöhnungseffekt des Medikaments zurückzuführen ist, der eine höhere Dosierung im Langzeitgebrauch erfordert. Die EPITOME-2-Studie deutet auf eine ähnliche kurzfristige Sicherheit und Wirksamkeit von Veletri ® bei PAH-Patienten hin, die stabil auf eine Flolan ® -Langzeittherapie eingestellt sind. Allerdings wurden deutlich höhere Dosen mit einem Mittelwert von 29,9 ± 15,1 ng/kg/min zu Studienbeginn und 30,2 ± 15,0 ng/kg/min nach drei Monaten verwendet [
7
]. In der kontrollierten 12-wöchigen Studie bei PAH wurde die Dosis von einer mittleren Anfangsdosis von 2,2 ng/kg/min erhöht. Während der ersten 7 Behandlungstage wurde die Dosis täglich auf eine mittlere Dosis von 4,1 ng/kg/min am 7. Behandlungstag erhöht. Am Ende von Woche 12 betrug die mittlere Dosis 11,2 ng/kg/min. Die mittlere schrittweise Erhöhung betrug 2–3 ng/kg/min alle 3 Wochen.Das Sicherheitsprofil in unserer Studie war mit Daten in der Literatur vergleichbar, wobei Übelkeit, Durchfall, Hitzewallungen und Kopfschmerzen zu den häufigsten unerwünschten Ereignissen im Zusammenhang mit dem Medikament gehörten [
2
].In Bezug auf die klinische Wirksamkeit gab es keine allgemeine Tendenz zur Verbesserung der gemessenen Parameter zur Beurteilung der körperlichen Leistungsfähigkeit (6MWD, Borg-Skala), der Lebensqualität (Kurzform-Gesundheitsbefragung 36; SF-36), der Hämodynamik, der rechten Herzgröße und Funktion sowie Lungenfunktion. Auch die WHO-FC blieb über den Untersuchungszeitraum stabil.Obwohl in dieser Studie keine klinisch relevante Verbesserung beobachtet wurde, blieben die meisten Patienten unter der Veletri® - Therapie stabil. In der Literatur wurde bereits über einen stabilen klinischen Verlauf unter Veletri ® -Behandlung berichtet, dies wurde jedoch bei Patienten beobachtet, die von Epoprostenol GM auf Epoprostenol AS umgestellt wurden [
7
,
15
,
16
]. Patienten, die von Epoprostenol GM auf Epoprostenol AS umgestellt wurden, berichteten von einem verbesserten Behandlungskomfort [
17
], während sich andere Parameter der Lebensqualität nicht veränderten. Obwohl in dieser Studie keine Änderung des Behandlungsschemas vorgenommen wurde, verbesserte sich die psychische Gesundheit bei zehn von fünfzehn Patienten.Bei Patienten mit neu begonnener Behandlung mit Epoprostenol umfassen die Behandlungseffekte eine Verbesserung der Hämodynamik [
5
,
18
,
19
], eine Verbesserung der körperlichen Belastbarkeit [
5
,
20
,
21
] und eine Verbesserung der Lebensqualität [
5
,
22
]. In unserer Kohorte konnten diese Effekte nicht beobachtet werden. Dies könnte auf die Schwere der Erkrankung zu Studienbeginn zurückzuführen sein, da fast die Hälfte der Studienkohorte im Verlauf der Studie und der Nachbeobachtung starb. Möglicherweise war die verabreichte Dosis von Veletri ® zu niedrig. Alle verstorbenen Patienten hatten jedoch Grunderkrankungen, die auf ein höheres Sterblichkeitsrisiko zurückzuführen sein könnten.Die Mehrheit der PH-bedingten UE (vier von sechs) wurde innerhalb von 3 Monaten nach Therapiebeginn beobachtet (Mittelwert: 120,2 ± 87,39 Tage, 95 %-KI: 28,6–211, und besserte sich im Laufe des Verlaufs, mit Ausnahme eines Todesfalls 19 Tage danach Veletri® - Initiation.Stärken und Grenzen der StudieDiese Studie wurde entwickelt, um Erkenntnisse über die klinische Praxis, Sicherheit, Verträglichkeit und den klinischen Verlauf während der Behandlung mit Veletri ® zu gewinnen . Ein beobachtendes Design ist vorzuziehen, wenn man darauf abzielt, Daten aus der realen Welt zu sammeln.Die Ergebnisse dieser Studie müssen unter Berücksichtigung ihrer potenziellen Einschränkungen aufgrund der geringen Anzahl von Teilnehmern und ihres nicht-interventionellen Designs betrachtet werden, das anfällig für Verluste bei der Nachverfolgung oder Abbrüche ist, die sich auf die Ergebnisse sowie die Signifikanz auswirken. Behandlungseffekte können aufgrund fehlender Werte von Patienten, die sich nicht bei jedem Besuch allen geplanten Untersuchungen unterzogen haben, möglicherweise nicht genau abgeschätzt werden. Darüber hinaus übersprang ein Patient die 3-monatige Nachbeobachtung, ein anderer Patient brach die Studie nach 3-monatiger Nachbeobachtung ab und drei Patienten starben während des 24-wöchigen Studienzeitraums aufgrund von Ursachen, die wahrscheinlich nicht mit iv Epoprostenol zusammenhängen. Insbesondere Änderungen der Endpunkte der Rechtsherzkatheterisierung konnten nicht zuverlässig bestimmt werden, da sich nur drei Patienten dieser Untersuchung zum 6-Monats-Follow-up unterzogen.Möglicherweise wurden Dosiserhöhungen zu niedrig durchgeführt. Während des stationären Aufenthaltes wurde Epoprostenol mit 2 ng/kg/min begonnen und alle 24 h um 1–2 ng/kg/min gesteigert, bis nach 5 Tagen eine Dosis von 10 ng/kg/min erreicht war (in Krankenhaus). Danach sollte die Dosis alle 1–2 Wochen um 1–2 ng/kg/min erhöht werden, um nach 6–8 Wochen 16 ng/kg/min zu erreichen. Bei den meisten Patienten haben wir diese Dosierung nicht erreicht. Möglicherweise haben wir nach dem stationären Therapiebeginn die Dosierung nicht ausreichend erhöht.Ziel der Studie war es, die klinische Erfahrung und das Wissen zu erweitern. Die Dosistitration ist ein wichtiger Teil, der in zukünftigen Anwendungen verbessert werden könnte und den wir vom PAH-Zentrum im Hôpital Bicêtre in Frankreich gelernt haben.Eine weitere Einschränkung dieser Studie ist der monozentrische Charakter dieser Studie. Daher kann ein Selektionsbias bei der Patientenrekrutierung nicht ausgeschlossen werden. Zudem war diese Studie als einarmige Open-Label-Studie geplant, weshalb eine Kontrollgruppe zum direkten Vergleich fehlt.Darüber hinaus kann die Verwendung von Fragebögen zu einer Verzerrung geführt haben, da Personen je nach Allgemeinzustand möglicherweise nicht gleichermaßen in der Lage sind, einen Fragebogen auszufüllen, was sich wiederum auf die aufgezeichneten Ergebnisse auswirkt.Im Allgemeinen war der Beobachtungszeitraum dieser Studie nicht lang genug, um die langfristigen Behandlungseffekte und das Überleben bei PAH-Patienten zuverlässig zu bestimmen. Weitere Studien mit einer größeren Teilnehmerzahl und einem verlängerten Überlebenszeitraum sind daher notwendig, um Titration, Dosierung und Dosiseskalation zu optimieren und die Wirksamkeit in Bezug auf das (transplantationsfreie) Überleben bei Patienten mit PAH zu bestimmen, um letztendlich eine Standardisierung zu leiten der Langzeittherapie.SchlussfolgerungenDie Studie zeigt, dass die Behandlung mit Epoprostenol (Veletri ® ) keine neuen Sicherheits- oder Verträglichkeitsaspekte ergab und bei den meisten Patienten durchführbar war. Die Überlebensrate war bei diesen schwer beeinträchtigten Patienten wie erwartet. In zukünftigen Anwendungen/Studien sollte der Up-Titrationsprozess auf höhere Epoprostenol-Dosierungen beim Auftreten von Nebenwirkungen drängen, da das Erreichen einer hohen und wirksamen Dosierung entscheidend für den klinischen Nutzen der Patienten ist.
Wood Units) wurden aufgenommen. Bei der Mehrheit war idiopathische pulmonale arterielle Hypertonie (IPAH) (66,6 %) oder PAH im Zusammenhang mit einer Bindegewebserkrankung (20 %, Tabelle
1
) diagnostiziert worden. Die mittlere Zeit von der Erstdiagnose bis zum Ausgangswert betrug 45,4 ± 23,7 Monate. Zu Beginn der Studie erhielten die meisten Patienten eine duale orale Kombinationsbehandlung (n = 14) mit PH-gerichteter Medikation in Übereinstimmung mit den Eignungskriterien (Tabelle
1
). Vier Patienten erhielten vor der Umstellung auf Veletri ® zusätzliche Prostazyklin-Analoga (inhalatives Iloprost n = 2, intravenöses Iloprost n = 1, subkutanes Treprostinil n = 1 ) . Patienten, die zuvor andere Prostacyclinanaloga wie Treprostinil oder Iloprost erhalten hatten, wurden auf Epoprostenol AS (Veletri ® ) umgestellt. Gründe für die Umstellung auf Veletri ® waren unzureichende Wirksamkeit bei den beiden Patienten mit inhalativem Iloprost und Probleme mit der Arzneimittelversorgung bei dem Patienten mit iv Iloprost. Der Patient, der zuvor Treprostinil erhielt, hatte wiederholt lokale Infektionen in der Nähe der Applikationsstelle und wurde daher auf Veletri ® umgestellt . Prostanoid-Behandlungen wurden über 3 Tage reduziert, um Veletri zu erhöhen® .Nach dem COMPERA-Ansatz befanden sich 14 Patienten in der mittleren Risikogruppe und einer in der Hochrisikogruppe. Sieben Patienten wiesen einen REVEAL-Risiko-Score ≥ 10 auf.Von 15 eingeschlossenen Patienten starben sieben Patienten und drei wurden während der Nachbeobachtungszeit für eine Lungentransplantation gelistet. Drei Patienten starben innerhalb des Studienzeitraums von 6 Monaten und vier Patienten starben während des Überlebens-Follow-up. Drei Patienten wurden für eine Lungentransplantation während der Überlebens-Follow-up-Periode aufgelistet.Elf Patienten beendeten die Studie (Behandlungsabbruch n = 1, Tod n = 3, Abb.
1
).Während der Studiendauer von 6 Monaten starben zwei Patienten einen Monat nach Behandlungsbeginn, einer 2½ Monate nach Behandlungsbeginn. Die Todesfälle waren auf multiples Organversagen aufgrund von Krebs im Endstadium (n = 2) und auf Rechtsherzversagen zurückzuführen. Abhängig von ihrem Allgemeinzustand wurden nicht alle Patienten bei jedem Studienbesuch allen geplanten Untersuchungen unterzogen. Ein Patient brach die Behandlung nach drei Monaten wegen Medikamentenunverträglichkeit und fehlendem Ansprechen auf die Behandlung mit gleichzeitiger Verschlechterung des PH ab. Dieser Patient starb etwa 3 Monate später. Ein Patient übersprang den Nachsorgetermin nach 3 Monaten und nahm am 6-Monats-Termin teil.Verwendung von Veletri®Veletri ® wurde im Krankenhaus über 7 ± 4 (1–12, Median 7) Tage titriert. Die Patienten wurden mit einer mittleren Maximaldosis von 7,9 ± 3,9 (3,4–20, Median 7,5) ng/kg/min behandelt, die meisten (12/15, 80 %) mit einer Dosis zwischen 5 und 10 ng/kg/min. Ein Patient, der eine deutlich höhere Dosierung von Veletri ® von 20 ng/kg/min erhielt, wurde zuvor mit Iloprost iv behandelt und konnte daher schneller und auf eine höhere verträgliche Dosis titriert werden (Abb.
2
). Weitere Dosierungen reichten von einer Höchstdosis von 3,4 ng/kg/min bis zu einer Höchstdosis von 10,4 ng/kg/min.Abb. 2
und besserte sich im Laufe des Verlaufs, mit Ausnahme eines Todesfalls 19 Tage danach Veletri® - Initiation.Stärken und Grenzen der StudieDiese Studie wurde entwickelt, um Erkenntnisse über die klinische Praxis, Sicherheit, Verträglichkeit und den klinischen Verlauf während der Behandlung mit Veletri ® zu gewinnen . Ein beobachtendes Design ist vorzuziehen, wenn man darauf abzielt, Daten aus der realen Welt zu sammeln.Die Ergebnisse dieser Studie müssen unter Berücksichtigung ihrer potenziellen Einschränkungen aufgrund der geringen Anzahl von Teilnehmern und ihres nicht-interventionellen Designs betrachtet werden, das anfällig für Verluste bei der Nachverfolgung oder Abbrüche ist, die sich auf die Ergebnisse sowie die Signifikanz auswirken. Behandlungseffekte können aufgrund fehlender Werte von Patienten, die sich nicht bei jedem Besuch allen geplanten Untersuchungen unterzogen haben, möglicherweise nicht genau abgeschätzt werden. Darüber hinaus übersprang ein Patient die 3-monatige Nachbeobachtung, ein anderer Patient brach die Studie nach 3-monatiger Nachbeobachtung ab und drei Patienten starben während des 24-wöchigen Studienzeitraums aufgrund von Ursachen, die wahrscheinlich nicht mit iv Epoprostenol zusammenhängen. Insbesondere Änderungen der Endpunkte der Rechtsherzkatheterisierung konnten nicht zuverlässig bestimmt werden, da sich nur drei Patienten dieser Untersuchung zum 6-Monats-Follow-up unterzogen.Möglicherweise wurden Dosiserhöhungen zu niedrig durchgeführt. Während des stationären Aufenthaltes wurde Epoprostenol mit 2 ng/kg/min begonnen und alle 24 h um 1–2 ng/kg/min gesteigert, bis nach 5 Tagen eine Dosis von 10 ng/kg/min erreicht war (in Krankenhaus). Danach sollte die Dosis alle 1–2 Wochen um 1–2 ng/kg/min erhöht werden, um nach 6–8 Wochen 16 ng/kg/min zu erreichen. Bei den meisten Patienten haben wir diese Dosierung nicht erreicht. Möglicherweise haben wir nach dem stationären Therapiebeginn die Dosierung nicht ausreichend erhöht.Ziel der Studie war es, die klinische Erfahrung und das Wissen zu erweitern. Die Dosistitration ist ein wichtiger Teil, der in zukünftigen Anwendungen verbessert werden könnte und den wir vom PAH-Zentrum im Hôpital Bicêtre in Frankreich gelernt haben.Eine weitere Einschränkung dieser Studie ist der monozentrische Charakter dieser Studie. Daher kann ein Selektionsbias bei der Patientenrekrutierung nicht ausgeschlossen werden. Zudem war diese Studie als einarmige Open-Label-Studie geplant, weshalb eine Kontrollgruppe zum direkten Vergleich fehlt.Darüber hinaus kann die Verwendung von Fragebögen zu einer Verzerrung geführt haben, da Personen je nach Allgemeinzustand möglicherweise nicht gleichermaßen in der Lage sind, einen Fragebogen auszufüllen, was sich wiederum auf die aufgezeichneten Ergebnisse auswirkt.Im Allgemeinen war der Beobachtungszeitraum dieser Studie nicht lang genug, um die langfristigen Behandlungseffekte und das Überleben bei PAH-Patienten zuverlässig zu bestimmen. Weitere Studien mit einer größeren Teilnehmerzahl und einem verlängerten Überlebenszeitraum sind daher notwendig, um Titration, Dosierung und Dosiseskalation zu optimieren und die Wirksamkeit in Bezug auf das (transplantationsfreie) Überleben bei Patienten mit PAH zu bestimmen, um letztendlich eine Standardisierung zu leiten der Langzeittherapie.SchlussfolgerungenDie Studie zeigt, dass die Behandlung mit Epoprostenol (Veletri ® ) keine neuen Sicherheits- oder Verträglichkeitsaspekte ergab und bei den meisten Patienten durchführbar war. Die Überlebensrate war bei diesen schwer beeinträchtigten Patienten wie erwartet. In zukünftigen Anwendungen/Studien sollte der Up-Titrationsprozess auf höhere Epoprostenol-Dosierungen beim Auftreten von Nebenwirkungen drängen, da das Erreichen einer hohen und wirksamen Dosierung entscheidend für den klinischen Nutzen der Patienten ist. OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.