
- Beiträge: 1757
Sidebar
- Forum
- PH-Forum
- Forschung und Wissen
- Sotatercept-Analogon unterdrückt die Entzündung, um die experimentelle PAH
Sotatercept-Analogon unterdrückt die Entzündung, um die experimentelle PAH
23 Mai 2022 16:07 #1474
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Sotatercept-Analogon unterdrückt die Entzündung, um die experimentelle PAH wurde erstellt von danny
www.nature.com/articles/s41598-022-11435-xDas Sotatercept-Analogon unterdrückt die Entzündung, um die experimentelle pulmonale arterielle Hypertonie umzukehren
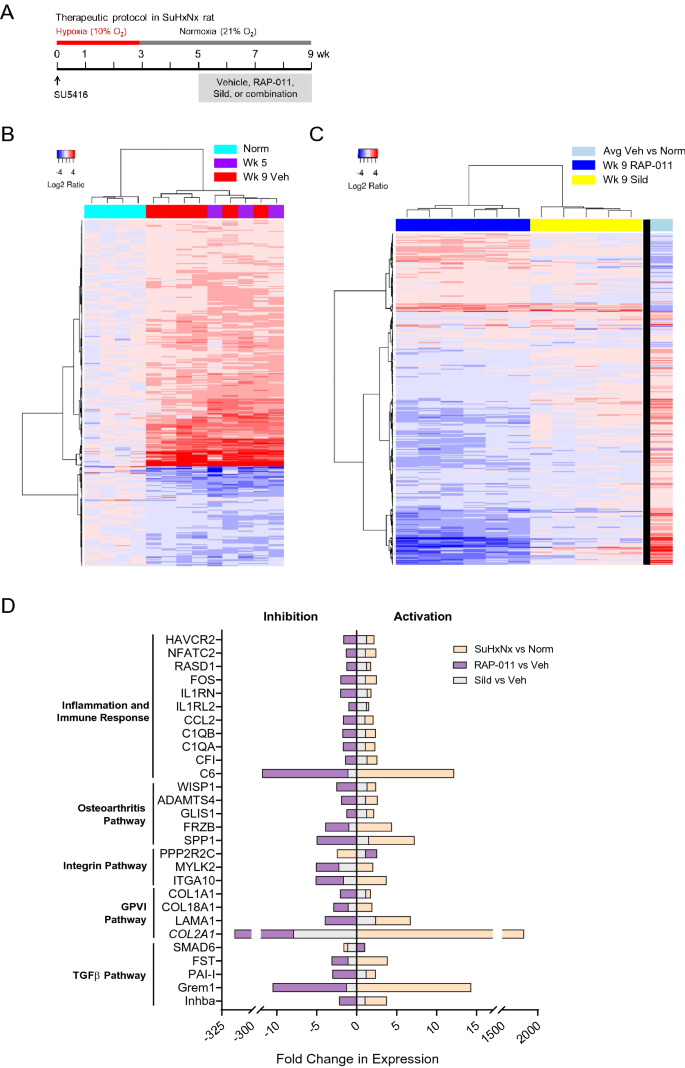 Die therapeutische Behandlung mit ActRIIA-Fc normalisiert weitgehend die pulmonale Genexpression bei schwerer experimenteller PAH. ( A ) Experimenteller Ansatz zur Bewertung der therapeutischen Wirkungen von RAP-011 in einem Sugen-Hypoxie-Normoxie (SuHxNx)-Rattenmodell für schwere PAH. Ratten wurden am Tag 0 mit einer Einzeldosis von SU5416 (20 mg/kg) behandelt und 3 Wochen lang einer normobaren Hypoxie (10 % O 2 ) ausgesetzt, gefolgt von 6 Wochen Normoxie, um ein Fortschreiten der Krankheit zu ermöglichen. Ratten wurden zusätzlich mit RAP-011 (2,5 mg/kg, sc, zweimal wöchentlich), Sildenafil (30 mg/kg, po, zweimal täglich), einer Kombinationstherapie mit RAP-011 und Sildenafil oder Vehikel (PBS) für 4 Wochen behandelt ab Woche 5 nach SU5416. ( B) Wärmekarte von differentiell exprimierten Genen (DEGs) in der Lunge von unbehandelten SuHxNx-Ratten in Woche 5 (Woche 5) und mit Vehikel behandelten SuHxNx-Ratten in Woche 9 (Woche 9 Veh), jeweils verglichen mit normal (Norm). Gene wurden unter Verwendung der Ward-Methode geclustert. ( C ) Wärmekarte von DEGs in Woche 9 in der Lunge von mit RAP-011 oder Sildenafil (Sild) behandelten SuHxNx-Ratten, jeweils verglichen mit einem normalisierten Durchschnitt von mit Vehikel behandelten SuHxNx-Ratten in Woche 9 (rechte Spalte). ( D ) IPA-basierte Klassifizierung ausgewählter Gene, die in Woche 9 in der Lunge von SuHxNx-Ratten, die mit Vehikel, RAP-011 oder Sildenafil behandelt wurden, eine signifikante unterschiedliche Expression zeigten.
Um die Voraussetzungen für die Expressionsprofilierung in der Lunge zu schaffen, bestätigten wir, dass hämodynamische Parameter, einschließlich des systolischen RV-Drucks (RVSP) und des gesamten pulmonalen Widerstandsindex (TPRI), bei unbehandelten SuHxNx-Ratten zu Beginn der therapeutischen Behandlung (Woche 5) und während dieser Dauer signifikant erhöht waren Woche 9 (ergänzende Abb.
1
). Eine verzögerte Behandlung mit RAP-011, beginnend in Woche 5, verbesserte (umgekehrte) hämodynamische Defizite bis Woche 9 deutlich im Vergleich zu mit Vehikel behandelten SuHxNx-Ratten (ergänzende
1
). Wir haben bestätigt, dass die Behandlung mit RAP-011 eine signifikant größere Verbesserung bewirkte als Sildenafil (ergänzende Abb.
1
), ein herkömmliches gefäßerweiterndes Mittel, das üblicherweise als Erst- oder Zweitlinientherapie bei PAH verwendet wird. Die Lungenhistologie bei unbehandelten SuHxNx-Ratten in Woche 5 und in Woche 9 mit Vehikel behandelten SuHxNx-Ratten zeigte eine signifikant erhöhte Häufigkeit umgestalteter und verschlossener Arterien, was mit den vorliegenden hämodynamischen Ergebnissen übereinstimmt (ergänzende
1
), und bestätigte unsere früheren Beobachtungen, dass die Behandlung mit RAP-011 eine Regression der Gefäße verursacht Remodeling effektiver als die Therapie mit einem Standard-Vasodilatator
28
.Als nächstes identifizierten wir Gene, die in SuHxNx-Rattenlunge im Vergleich zu normaler Rattenlunge differentiell exprimiert wurden. Unter Verwendung der hierarchischen Clusteranalyse von RNA-seq-Daten identifizierten wir 345 differentiell exprimierte Gene (DEGs) mit einem Fold-Change ≥ 1,5 und einem angepassten p-Wert ≤ 0,05 sowohl in Woche 5 als auch in Woche 9. Von diesen 345 DEGs waren 248 hochreguliert und 97 wurden in erkrankter Rattenlunge im Vergleich zu normalem Gewebe herunterreguliert (Abb.
1
Die therapeutische Behandlung mit ActRIIA-Fc normalisiert weitgehend die pulmonale Genexpression bei schwerer experimenteller PAH. ( A ) Experimenteller Ansatz zur Bewertung der therapeutischen Wirkungen von RAP-011 in einem Sugen-Hypoxie-Normoxie (SuHxNx)-Rattenmodell für schwere PAH. Ratten wurden am Tag 0 mit einer Einzeldosis von SU5416 (20 mg/kg) behandelt und 3 Wochen lang einer normobaren Hypoxie (10 % O 2 ) ausgesetzt, gefolgt von 6 Wochen Normoxie, um ein Fortschreiten der Krankheit zu ermöglichen. Ratten wurden zusätzlich mit RAP-011 (2,5 mg/kg, sc, zweimal wöchentlich), Sildenafil (30 mg/kg, po, zweimal täglich), einer Kombinationstherapie mit RAP-011 und Sildenafil oder Vehikel (PBS) für 4 Wochen behandelt ab Woche 5 nach SU5416. ( B) Wärmekarte von differentiell exprimierten Genen (DEGs) in der Lunge von unbehandelten SuHxNx-Ratten in Woche 5 (Woche 5) und mit Vehikel behandelten SuHxNx-Ratten in Woche 9 (Woche 9 Veh), jeweils verglichen mit normal (Norm). Gene wurden unter Verwendung der Ward-Methode geclustert. ( C ) Wärmekarte von DEGs in Woche 9 in der Lunge von mit RAP-011 oder Sildenafil (Sild) behandelten SuHxNx-Ratten, jeweils verglichen mit einem normalisierten Durchschnitt von mit Vehikel behandelten SuHxNx-Ratten in Woche 9 (rechte Spalte). ( D ) IPA-basierte Klassifizierung ausgewählter Gene, die in Woche 9 in der Lunge von SuHxNx-Ratten, die mit Vehikel, RAP-011 oder Sildenafil behandelt wurden, eine signifikante unterschiedliche Expression zeigten.
Um die Voraussetzungen für die Expressionsprofilierung in der Lunge zu schaffen, bestätigten wir, dass hämodynamische Parameter, einschließlich des systolischen RV-Drucks (RVSP) und des gesamten pulmonalen Widerstandsindex (TPRI), bei unbehandelten SuHxNx-Ratten zu Beginn der therapeutischen Behandlung (Woche 5) und während dieser Dauer signifikant erhöht waren Woche 9 (ergänzende Abb.
1
). Eine verzögerte Behandlung mit RAP-011, beginnend in Woche 5, verbesserte (umgekehrte) hämodynamische Defizite bis Woche 9 deutlich im Vergleich zu mit Vehikel behandelten SuHxNx-Ratten (ergänzende
1
). Wir haben bestätigt, dass die Behandlung mit RAP-011 eine signifikant größere Verbesserung bewirkte als Sildenafil (ergänzende Abb.
1
), ein herkömmliches gefäßerweiterndes Mittel, das üblicherweise als Erst- oder Zweitlinientherapie bei PAH verwendet wird. Die Lungenhistologie bei unbehandelten SuHxNx-Ratten in Woche 5 und in Woche 9 mit Vehikel behandelten SuHxNx-Ratten zeigte eine signifikant erhöhte Häufigkeit umgestalteter und verschlossener Arterien, was mit den vorliegenden hämodynamischen Ergebnissen übereinstimmt (ergänzende
1
), und bestätigte unsere früheren Beobachtungen, dass die Behandlung mit RAP-011 eine Regression der Gefäße verursacht Remodeling effektiver als die Therapie mit einem Standard-Vasodilatator
28
.Als nächstes identifizierten wir Gene, die in SuHxNx-Rattenlunge im Vergleich zu normaler Rattenlunge differentiell exprimiert wurden. Unter Verwendung der hierarchischen Clusteranalyse von RNA-seq-Daten identifizierten wir 345 differentiell exprimierte Gene (DEGs) mit einem Fold-Change ≥ 1,5 und einem angepassten p-Wert ≤ 0,05 sowohl in Woche 5 als auch in Woche 9. Von diesen 345 DEGs waren 248 hochreguliert und 97 wurden in erkrankter Rattenlunge im Vergleich zu normalem Gewebe herunterreguliert (Abb.
1
") . Die therapeutische Behandlung von SuHxNx-Ratten mit RAP-011 von Woche 5 bis Woche 9 übte eine robuste normalisierende Wirkung auf dieses pathologische Genexpressionsprofil aus (Abb.
1
C). Bis Woche 9 normalisierte die Behandlung mit RAP-011 die Expression von 207 von 248 (84 %) hochregulierten DEGs und 69 von 97 (71 %) herunterregulierten DEGs.
Im Gegensatz dazu veränderte die therapeutische Behandlung von SuHxNx
-Ratten mit Sildenafil die Expression von nur 27 von insgesamt 345 (8 %) DEGs ( 1C ). Die Hauptkomponentenanalyse ergab, dass Lungengewebe von mit RAP-011 behandelten SuHxNx-Ratten ein Genexpressionsprofil aufwies, das global normalem Gewebe ähnelte, während das Profil der mit Sildenafil behandelten SuHxNx-Rattenlungen in Woche 5 stärker dem der unbehandelten SuHxNx-Rattenlungen ähnelte (ergänzende Abb
2
_). Diese Ergebnisse weisen darauf hin, dass die therapeutische Behandlung mit RAP-011 eine deutliche korrigierende Wirkung auf das globale pathologische Genexpressionsprofil bei schwerer experimenteller PAH ausübt, die von der Behandlung mit einem Standard-Vasodilatator, der derzeit für die PAH-Therapie verfügbar ist, nicht erreicht wird.Anschließend verwendeten wir die Ingenuity Pathway Analysis (IPA), um dysregulierte Signalwege und potenzielle vorgeschaltete Regulatoren zu identifizieren, die mit allen DEGs (definiert durch den angepassten p-Wert < 0,001) assoziiert sind, basierend auf einem Vergleich des Lungengewebes in unbehandelten SuHxNx-Ratten sowohl in Woche 5 als auch in Woche 9 mit Lungengewebe bei normalen Ratten. Diese Analyse identifizierte 58 Signalwege, die in erkrankten Lungen sowohl in Woche 5 als auch in Woche 9 im Vergleich zu normalem Gewebe signifikant fehlreguliert waren. Gemäß der Rangfolge nach der Fisher-Methode umfassen die wichtigsten kanonischen Wege diejenigen, die Endothel- und Gefäßverletzungsreaktionen vermitteln (Gerinnungs-, Prothrombinaktivierungs- und Glykoprotein-VI-Wege); Entzündung und Immunantwort (Komplement, dendritische Zellreifung, Mustererkennung, Interleukin-10 und angeborene und adaptive Immunantwortwege); und TGF-β-Signalisierung (Tabelle
1
), die alle mit dem Fortschreiten der PAH bei Patienten oder präklinischen Modellen oder beidem in Verbindung gebracht wurden
2
,
4
. Interessanterweise war der wichtigste vorgeschaltete Regulator, der durch diese Analyse identifiziert wurde, der Tumornekrosefaktor (TNF), ein Schlüsselregulator von Entzündungs- und Immunantworten, der auch die Expression von BMPRII hemmt
37
. Andere hochrangige Upstream-Regulatoren wie VCAN, EPHB1, EGLN1 und TSC2 sind dafür bekannt, dass sie die Zellproliferation und -migration regulieren. Weitere Kandidaten für vorgeschaltete Regulatoren, die von IPA identifiziert wurden, umfassen TGFBR2 und Gene, die am Mitogen-aktivierten Proteinkinase-Weg beteiligt sind (ERK, p38MAPK und MAP2k1), gut charakterisierte Regulatoren von Entzündung und Zellproliferation (Tabelle
1
). Insgesamt zeigen die Signalwege und vorgeschalteten Regulatoren, die durch diese Analyse in der SuHxNx-Lungenpathologie involviert sind, einen Phänotyp einer hochgradig aktivierten Entzündung und Proliferation.Tabelle 1 Die am besten eingestuften Signalwege und vorgeschalteten Regulatoren gemäß Ingenuity Pathway Analysis in Lungen eines SuHxNx-Rattenmodells schwerer PAH.
Übereinstimmung bei abweichenden Genexpressionsprofilen zwischen einem Rattenmodell mit schwerer angioobliterativer PAH und PAH-PatientenAls nächstes untersuchten wir die potenzielle Relevanz dieser fehlregulierten Signalwege für die PAH-Pathogenese bei Patienten. Öffentlich zugängliche menschliche Transkriptomdaten, die von 58 PAH-Lungen und 25 Kontrolllungen (
www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261
) gesammelt wurden, wurden IPA unterzogen, um signifikant fehlregulierte zu identifizieren Signalwege, die dann mit den in SuHxNx-Rattenlunge identifizierten Signalwegen verglichen wurden (Tabelle
1
). In Übereinstimmung mit veröffentlichten Humandaten umfassten die hier im Lungengewebe von PAH-Patienten identifizierten Signalwege an erster Stelle solche, die G-Protein-gekoppelte Rezeptoren beinhalten, und mehrere Signalwege, die mit Entzündungen und Immunantworten verbunden sind. Wichtig ist, dass IPA 18 Signalwege im Zusammenhang mit Entzündungs- und Immunantworten identifizierte, die im Lungengewebe von PAH-Patienten und dem SuHxNx-Rattenmodell für schwere PAH signifikant fehlreguliert sind (Tabelle
2
). Unter anderen solchen Signalwegen sind jene, die TGF-β- und BMP-Signalgebung vermitteln. Diese vom IPA erhaltenen Ergebnisse weisen auf eine Übereinstimmung in der fehlregulierten Genexpression zwischen dem SuHxNx-Rattenmodell und dem Lungengewebe von PAH-Patienten hin und liefern einen bestätigenden Beweis dafür, dass die hier verwendete SuHxNx-Ratte ein robustes Modell für menschliche PAH ist, insbesondere in Bezug auf ihre überschwängliche entzündliche Genexpressionssignatur.Tabelle 2 Häufigste Wege in der Lunge von SuHxNx-Ratten und PAH-Patienten.
ActRIIA-Fc zielt in verschiedenen PH-Modellen auf pulmonale Entzündungsmarker und Makrophageninfiltration abUm die Auswirkungen einer therapeutischen Behandlung mit entweder RAP-011 oder Sildenafil auf fehlregulierte Signalwege in der SuHxNx-Rattenlunge zu beurteilen, haben wir die relativen Expressionsniveaus von DEGs bestimmt, die mit diesen Signalwegen assoziiert sind. Die therapeutische Behandlung mit RAP-011 kehrte die anomale Expression vieler Gene in der Lunge um, die ansonsten im Zustand der SuHxNx-Krankheit aktiviert wurden (Abb. 1D
)
. Gene, die als Ergebnis der RAP-011-Behandlung differentiell exprimiert wurden, schlossen prominent jene in Signalwegen ein, die mit entzündlichen und abweichenden Immunantworten assoziiert sind ( 1D
)
. Andere DEGs sind mit dem Signalweg der TGF-β-Superfamilie assoziiert, insbesondere Grem1 (Gremlin-1), ein endogener BMP-Antagonist, der an der Proliferation von Endothelzellen und PAH beteiligt ist
38
. Mit der RAP-011-Behandlung verbundene Expressionsänderungen waren in den Fällen von Col2A1 , C6 und Grem1 besonders ausgeprägt . Im Gegensatz zu den auffälligen korrigierenden Wirkungen der RAP-011-Behandlung führte die therapeutische Behandlung mit Sildenafil zu begrenzten Veränderungen in der Expression von krankheitsassoziierten Genen (Abb.
1D
). Diese Ergebnisse liefern eine pulmonale Genexpressionssignatur, die den potenten therapeutischen Anti-Remodeling-Effekten von RAP-011 in diesem Modell schwerer PAH entspricht.Anschließend untersuchten wir die Auswirkungen der RAP-011-Therapie auf die Expression ausgewählter molekularer Entzündungs- und Immunmarker in der SuHxNx-Rattenlunge. Die PAH-Progression bei SuHxNx-Ratten war in Woche 9 mit einer signifikant erhöhten Expression von acht Schlüsselmarkern, einschließlich Il6 und Ccl2 , verbunden, und in jedem Fall normalisierte die therapeutische Behandlung mit RAP-011 – aber nicht Vehikel oder Sildenafil – ihre mRNA-Spiegel vollständig (Abb.
2
EIN). Wichtig ist, dass RAP-011 und Sildenafil in Kombination die Expression dieser Marker genauso effektiv normalisierten wie die RAP-011-Monotherapie (Abb. 2A
)
, was darauf hinweist, dass RAP-011 in diesem Modell einen robusten Nutzen bietet, selbst wenn es in Kombination mit einem Standard-Vasodilatator verwendet wird.Figur 2
. Die therapeutische Behandlung von SuHxNx-Ratten mit RAP-011 von Woche 5 bis Woche 9 übte eine robuste normalisierende Wirkung auf dieses pathologische Genexpressionsprofil aus (Abb.
1
C). Bis Woche 9 normalisierte die Behandlung mit RAP-011 die Expression von 207 von 248 (84 %) hochregulierten DEGs und 69 von 97 (71 %) herunterregulierten DEGs.
Im Gegensatz dazu veränderte die therapeutische Behandlung von SuHxNx
-Ratten mit Sildenafil die Expression von nur 27 von insgesamt 345 (8 %) DEGs ( 1C ). Die Hauptkomponentenanalyse ergab, dass Lungengewebe von mit RAP-011 behandelten SuHxNx-Ratten ein Genexpressionsprofil aufwies, das global normalem Gewebe ähnelte, während das Profil der mit Sildenafil behandelten SuHxNx-Rattenlungen in Woche 5 stärker dem der unbehandelten SuHxNx-Rattenlungen ähnelte (ergänzende Abb
2
_). Diese Ergebnisse weisen darauf hin, dass die therapeutische Behandlung mit RAP-011 eine deutliche korrigierende Wirkung auf das globale pathologische Genexpressionsprofil bei schwerer experimenteller PAH ausübt, die von der Behandlung mit einem Standard-Vasodilatator, der derzeit für die PAH-Therapie verfügbar ist, nicht erreicht wird.Anschließend verwendeten wir die Ingenuity Pathway Analysis (IPA), um dysregulierte Signalwege und potenzielle vorgeschaltete Regulatoren zu identifizieren, die mit allen DEGs (definiert durch den angepassten p-Wert < 0,001) assoziiert sind, basierend auf einem Vergleich des Lungengewebes in unbehandelten SuHxNx-Ratten sowohl in Woche 5 als auch in Woche 9 mit Lungengewebe bei normalen Ratten. Diese Analyse identifizierte 58 Signalwege, die in erkrankten Lungen sowohl in Woche 5 als auch in Woche 9 im Vergleich zu normalem Gewebe signifikant fehlreguliert waren. Gemäß der Rangfolge nach der Fisher-Methode umfassen die wichtigsten kanonischen Wege diejenigen, die Endothel- und Gefäßverletzungsreaktionen vermitteln (Gerinnungs-, Prothrombinaktivierungs- und Glykoprotein-VI-Wege); Entzündung und Immunantwort (Komplement, dendritische Zellreifung, Mustererkennung, Interleukin-10 und angeborene und adaptive Immunantwortwege); und TGF-β-Signalisierung (Tabelle
1
), die alle mit dem Fortschreiten der PAH bei Patienten oder präklinischen Modellen oder beidem in Verbindung gebracht wurden
2
,
4
. Interessanterweise war der wichtigste vorgeschaltete Regulator, der durch diese Analyse identifiziert wurde, der Tumornekrosefaktor (TNF), ein Schlüsselregulator von Entzündungs- und Immunantworten, der auch die Expression von BMPRII hemmt
37
. Andere hochrangige Upstream-Regulatoren wie VCAN, EPHB1, EGLN1 und TSC2 sind dafür bekannt, dass sie die Zellproliferation und -migration regulieren. Weitere Kandidaten für vorgeschaltete Regulatoren, die von IPA identifiziert wurden, umfassen TGFBR2 und Gene, die am Mitogen-aktivierten Proteinkinase-Weg beteiligt sind (ERK, p38MAPK und MAP2k1), gut charakterisierte Regulatoren von Entzündung und Zellproliferation (Tabelle
1
). Insgesamt zeigen die Signalwege und vorgeschalteten Regulatoren, die durch diese Analyse in der SuHxNx-Lungenpathologie involviert sind, einen Phänotyp einer hochgradig aktivierten Entzündung und Proliferation.Tabelle 1 Die am besten eingestuften Signalwege und vorgeschalteten Regulatoren gemäß Ingenuity Pathway Analysis in Lungen eines SuHxNx-Rattenmodells schwerer PAH.
Übereinstimmung bei abweichenden Genexpressionsprofilen zwischen einem Rattenmodell mit schwerer angioobliterativer PAH und PAH-PatientenAls nächstes untersuchten wir die potenzielle Relevanz dieser fehlregulierten Signalwege für die PAH-Pathogenese bei Patienten. Öffentlich zugängliche menschliche Transkriptomdaten, die von 58 PAH-Lungen und 25 Kontrolllungen (
www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261
) gesammelt wurden, wurden IPA unterzogen, um signifikant fehlregulierte zu identifizieren Signalwege, die dann mit den in SuHxNx-Rattenlunge identifizierten Signalwegen verglichen wurden (Tabelle
1
). In Übereinstimmung mit veröffentlichten Humandaten umfassten die hier im Lungengewebe von PAH-Patienten identifizierten Signalwege an erster Stelle solche, die G-Protein-gekoppelte Rezeptoren beinhalten, und mehrere Signalwege, die mit Entzündungen und Immunantworten verbunden sind. Wichtig ist, dass IPA 18 Signalwege im Zusammenhang mit Entzündungs- und Immunantworten identifizierte, die im Lungengewebe von PAH-Patienten und dem SuHxNx-Rattenmodell für schwere PAH signifikant fehlreguliert sind (Tabelle
2
). Unter anderen solchen Signalwegen sind jene, die TGF-β- und BMP-Signalgebung vermitteln. Diese vom IPA erhaltenen Ergebnisse weisen auf eine Übereinstimmung in der fehlregulierten Genexpression zwischen dem SuHxNx-Rattenmodell und dem Lungengewebe von PAH-Patienten hin und liefern einen bestätigenden Beweis dafür, dass die hier verwendete SuHxNx-Ratte ein robustes Modell für menschliche PAH ist, insbesondere in Bezug auf ihre überschwängliche entzündliche Genexpressionssignatur.Tabelle 2 Häufigste Wege in der Lunge von SuHxNx-Ratten und PAH-Patienten.
ActRIIA-Fc zielt in verschiedenen PH-Modellen auf pulmonale Entzündungsmarker und Makrophageninfiltration abUm die Auswirkungen einer therapeutischen Behandlung mit entweder RAP-011 oder Sildenafil auf fehlregulierte Signalwege in der SuHxNx-Rattenlunge zu beurteilen, haben wir die relativen Expressionsniveaus von DEGs bestimmt, die mit diesen Signalwegen assoziiert sind. Die therapeutische Behandlung mit RAP-011 kehrte die anomale Expression vieler Gene in der Lunge um, die ansonsten im Zustand der SuHxNx-Krankheit aktiviert wurden (Abb. 1D
)
. Gene, die als Ergebnis der RAP-011-Behandlung differentiell exprimiert wurden, schlossen prominent jene in Signalwegen ein, die mit entzündlichen und abweichenden Immunantworten assoziiert sind ( 1D
)
. Andere DEGs sind mit dem Signalweg der TGF-β-Superfamilie assoziiert, insbesondere Grem1 (Gremlin-1), ein endogener BMP-Antagonist, der an der Proliferation von Endothelzellen und PAH beteiligt ist
38
. Mit der RAP-011-Behandlung verbundene Expressionsänderungen waren in den Fällen von Col2A1 , C6 und Grem1 besonders ausgeprägt . Im Gegensatz zu den auffälligen korrigierenden Wirkungen der RAP-011-Behandlung führte die therapeutische Behandlung mit Sildenafil zu begrenzten Veränderungen in der Expression von krankheitsassoziierten Genen (Abb.
1D
). Diese Ergebnisse liefern eine pulmonale Genexpressionssignatur, die den potenten therapeutischen Anti-Remodeling-Effekten von RAP-011 in diesem Modell schwerer PAH entspricht.Anschließend untersuchten wir die Auswirkungen der RAP-011-Therapie auf die Expression ausgewählter molekularer Entzündungs- und Immunmarker in der SuHxNx-Rattenlunge. Die PAH-Progression bei SuHxNx-Ratten war in Woche 9 mit einer signifikant erhöhten Expression von acht Schlüsselmarkern, einschließlich Il6 und Ccl2 , verbunden, und in jedem Fall normalisierte die therapeutische Behandlung mit RAP-011 – aber nicht Vehikel oder Sildenafil – ihre mRNA-Spiegel vollständig (Abb.
2
EIN). Wichtig ist, dass RAP-011 und Sildenafil in Kombination die Expression dieser Marker genauso effektiv normalisierten wie die RAP-011-Monotherapie (Abb. 2A
)
, was darauf hinweist, dass RAP-011 in diesem Modell einen robusten Nutzen bietet, selbst wenn es in Kombination mit einem Standard-Vasodilatator verwendet wird.Figur 2
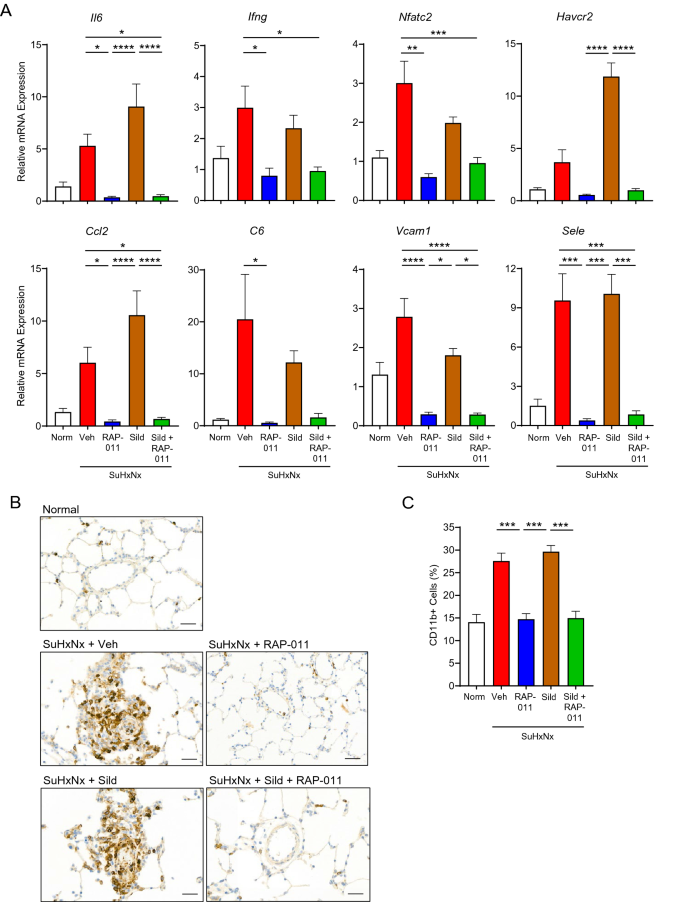 Die therapeutische Behandlung mit ActRIIA-Fc unterdrückt Lungenentzündungen und abweichende Immunantworten bei schwerer experimenteller PAH. ( A ) Niveaus von Il6- , Ifng- , Nfatc2- , Havcr2- , Ccl2- , C6- , Vcam1- und Sele - mRNA in der Lunge von normalen Ratten (Norm) oder SuHxNx-Ratten, die mit Vehikel (Veh), RAP-011, Sildenafil (Sild) behandelt wurden, oder eine Kombination aus Sildenafil und RAP-011. Die Daten sind Mittelwerte ± SEM (n = 6–9 Ratten pro Gruppe). ( B) Repräsentative Bilder von Lungenschnitten, die für den Makrophagenmarker CD11b immungefärbt wurden und deutliche Cluster von markierten perivaskulären Zellen bei schwerer experimenteller PAH nach Behandlung mit Vehikel oder Sildenafil, aber nicht mit RAP-011 zeigen. ( C ) Quantifizierung von CD11b-positiven Zellen in der Lunge basierend auf der Bewertung von 40 stark vergrößerten Feldern pro Ratte. Maßstabsleiste, 50 µm. Die Daten sind Mittelwerte ± SEM (n = 6–9 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-hoc-Test (* P < 0,05, ** P < 0,01, *** P < 0,001, **** P < 0,0001).
Wir untersuchten die Auswirkungen einer therapeutischen Behandlung mit RAP-011 auf die Expression von CD11b, einem Marker für aktivierte Alveolarmakrophagen – die vorherrschenden Immunzellen in den alveolären Lufträumen
39
. Wie durch Immunfärbung bestimmt, war die perivaskuläre CD11b + -Zellhäufigkeit in der Lunge von SuHxNx-Ratten in Woche 9 im Vergleich zu Werten in normalen Rattenlungen signifikant erhöht (
2
B, C). Wie bei anderen Immunmarkern blockierte die therapeutische Behandlung mit RAP-011 die pulmonale Infiltration von CD11b + -Zellen vollständig, während die Behandlung mit Vehikel oder Sildenafil keine Wirkung hatte (
2
B, C). RAP-011 normalisierte die Anzahl der pulmonalen CD11b+-Zellen vollständig, wenn es in Kombination mit Sildenafil verabreicht wurde (Abb.
2
B, C), was die entzündungshemmende Wirksamkeit von RAP-011 entweder als Monotherapie oder als Zusatztherapie in diesem Setting demonstriert. Wie durch fluoreszenzaktivierte Zellsortierung festgestellt wurde, blockierte die vorbeugende Behandlung mit RAP-011 in ähnlicher Weise die pulmonale Infiltration von CD11b + -Zellen bei mit Monocrotalin behandelten Ratten, einem weiteren etablierten Modell für induzierbaren PH
40
, während sie auch die Entwicklung von erhöhtem RVSP und RV-Hypertrophie verhinderte (ergänzende Abb
3
) . Diese Ergebnisse zeigen, dass die therapeutische Behandlung mit ActRIIA-Fc – im Gegensatz zur PAH-Standardtherapie mit Sildenafil – Entzündungen und perivaskuläre Monozyteninfiltration als wichtige Komponenten seiner Anti-Remodeling-Aktivität auf Gewebeebene in PH-Modellen stark hemmt.Liganden der Activin-Klasse tragen zur Makrophagenaktivierung und zum kardiopulmonalen Umbau beiAls nächstes untersuchten wir die Wirkungen von Liganden der Activin-Klasse auf die Expression von molekularen Markern der entzündlichen Makrophagenaktivierung in vitro. THP1-Zellen, eine humane Monozyten-Zelllinie, zeigten unterschiedliche Muster der Geninduktion, wenn sie Activin A, Activin B oder GDF11 ausgesetzt wurden (Fig. 3A
)
. Insbesondere erhöhte Aktivin A die Expression von Ccl2 , Il6 , Il1b und Tnf ; Aktivin B erhöhte selektiv Il6 ; und GDF11 erhöhte Ccl2 , Il6 und Il1b. Wie erwartet verursachte die Behandlung von THP1-Zellen mit einer Dreifachkombination aus Aktivin A, Aktivin B und GDF11 auch eine Hochregulierung von molekularen Entzündungsmarkern, und die gleichzeitige Behandlung mit einem menschlichen ActRIIA-Fc-Analogon (ACE-011) verhinderte diesen Effekt (ergänzende Abb
4
) . Diese Ergebnisse zeigen, dass Liganden der Activin-Klasse individuell unterschiedliche aktivierende Wirkungen auf Monozyten ausüben können, um einen proinflammatorischen Makrophagen-Phänotyp in vitro zu fördern.Figur 3
Die therapeutische Behandlung mit ActRIIA-Fc unterdrückt Lungenentzündungen und abweichende Immunantworten bei schwerer experimenteller PAH. ( A ) Niveaus von Il6- , Ifng- , Nfatc2- , Havcr2- , Ccl2- , C6- , Vcam1- und Sele - mRNA in der Lunge von normalen Ratten (Norm) oder SuHxNx-Ratten, die mit Vehikel (Veh), RAP-011, Sildenafil (Sild) behandelt wurden, oder eine Kombination aus Sildenafil und RAP-011. Die Daten sind Mittelwerte ± SEM (n = 6–9 Ratten pro Gruppe). ( B) Repräsentative Bilder von Lungenschnitten, die für den Makrophagenmarker CD11b immungefärbt wurden und deutliche Cluster von markierten perivaskulären Zellen bei schwerer experimenteller PAH nach Behandlung mit Vehikel oder Sildenafil, aber nicht mit RAP-011 zeigen. ( C ) Quantifizierung von CD11b-positiven Zellen in der Lunge basierend auf der Bewertung von 40 stark vergrößerten Feldern pro Ratte. Maßstabsleiste, 50 µm. Die Daten sind Mittelwerte ± SEM (n = 6–9 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-hoc-Test (* P < 0,05, ** P < 0,01, *** P < 0,001, **** P < 0,0001).
Wir untersuchten die Auswirkungen einer therapeutischen Behandlung mit RAP-011 auf die Expression von CD11b, einem Marker für aktivierte Alveolarmakrophagen – die vorherrschenden Immunzellen in den alveolären Lufträumen
39
. Wie durch Immunfärbung bestimmt, war die perivaskuläre CD11b + -Zellhäufigkeit in der Lunge von SuHxNx-Ratten in Woche 9 im Vergleich zu Werten in normalen Rattenlungen signifikant erhöht (
2
B, C). Wie bei anderen Immunmarkern blockierte die therapeutische Behandlung mit RAP-011 die pulmonale Infiltration von CD11b + -Zellen vollständig, während die Behandlung mit Vehikel oder Sildenafil keine Wirkung hatte (
2
B, C). RAP-011 normalisierte die Anzahl der pulmonalen CD11b+-Zellen vollständig, wenn es in Kombination mit Sildenafil verabreicht wurde (Abb.
2
B, C), was die entzündungshemmende Wirksamkeit von RAP-011 entweder als Monotherapie oder als Zusatztherapie in diesem Setting demonstriert. Wie durch fluoreszenzaktivierte Zellsortierung festgestellt wurde, blockierte die vorbeugende Behandlung mit RAP-011 in ähnlicher Weise die pulmonale Infiltration von CD11b + -Zellen bei mit Monocrotalin behandelten Ratten, einem weiteren etablierten Modell für induzierbaren PH
40
, während sie auch die Entwicklung von erhöhtem RVSP und RV-Hypertrophie verhinderte (ergänzende Abb
3
) . Diese Ergebnisse zeigen, dass die therapeutische Behandlung mit ActRIIA-Fc – im Gegensatz zur PAH-Standardtherapie mit Sildenafil – Entzündungen und perivaskuläre Monozyteninfiltration als wichtige Komponenten seiner Anti-Remodeling-Aktivität auf Gewebeebene in PH-Modellen stark hemmt.Liganden der Activin-Klasse tragen zur Makrophagenaktivierung und zum kardiopulmonalen Umbau beiAls nächstes untersuchten wir die Wirkungen von Liganden der Activin-Klasse auf die Expression von molekularen Markern der entzündlichen Makrophagenaktivierung in vitro. THP1-Zellen, eine humane Monozyten-Zelllinie, zeigten unterschiedliche Muster der Geninduktion, wenn sie Activin A, Activin B oder GDF11 ausgesetzt wurden (Fig. 3A
)
. Insbesondere erhöhte Aktivin A die Expression von Ccl2 , Il6 , Il1b und Tnf ; Aktivin B erhöhte selektiv Il6 ; und GDF11 erhöhte Ccl2 , Il6 und Il1b. Wie erwartet verursachte die Behandlung von THP1-Zellen mit einer Dreifachkombination aus Aktivin A, Aktivin B und GDF11 auch eine Hochregulierung von molekularen Entzündungsmarkern, und die gleichzeitige Behandlung mit einem menschlichen ActRIIA-Fc-Analogon (ACE-011) verhinderte diesen Effekt (ergänzende Abb
4
) . Diese Ergebnisse zeigen, dass Liganden der Activin-Klasse individuell unterschiedliche aktivierende Wirkungen auf Monozyten ausüben können, um einen proinflammatorischen Makrophagen-Phänotyp in vitro zu fördern.Figur 3
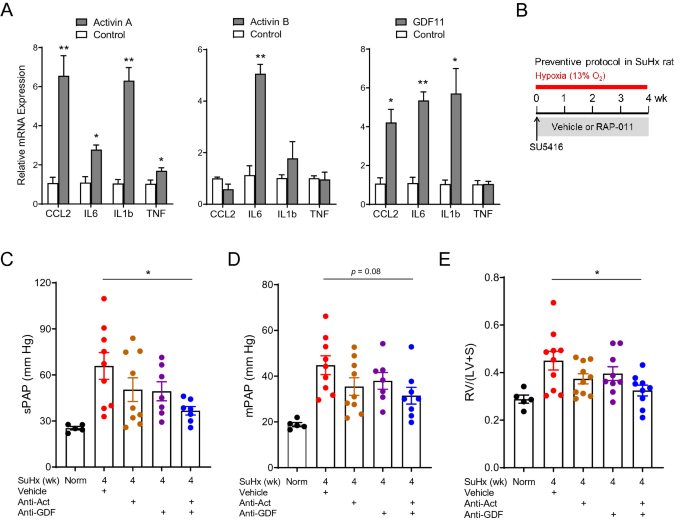 Mehrere Liganden tragen zur Makrophagenaktivierung in vitro und zum kardiopulmonalen Umbau in einem SuHx-Rattenmodell von PH bei. ( A ) Expression von Markern der Makrophagenaktivierung in THP-1-Monozyten in vitro ohne Behandlung (Kontrolle) oder nach Behandlung mit Aktivin A (5 ng/ml), Aktivin B (50 ng/ml) oder GDF11 (5 ng/ml ). Analyse durch ungepaarten t-Test (* P < 0,05, ** P < 0,01 vs. Kontrolle). ( B ) Experimenteller Ansatz, der zum Testen der Wirkungen der Multi-Liganden-Hemmung in einem SuHx-Rattenmodell von PH verwendet wurde. Ratten wurden mit einer Einzeldosis von SU5416 (20 mg/kg, sc) behandelt und einer normobaren Hypoxie (13 % O2) ausgesetzt) und zweimal wöchentlich subkutan behandelt mit separaten Antikörpern gegen Aktivin A und Aktivin B (Anti-Act, jeweils 10 mg/kg), einem Antikörper mit doppelter Spezifität für GDF8 und GDF11 (Anti-GDF, 10 mg/kg), kombiniertem Anti – Act und Anti-GDF oder Vehikel (PBS) für 4 Wochen, beginnend einen Tag nach SU5416. ( C ) sPAP, ( D ) mPAP und ( E ) Fulton-Index. Die Daten sind Mittelwerte ± SEM (n = 5–9 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test (* P < 0,05, ** P < 0,01, **** P < 0,0001).
Wir verwendeten neutralisierende Antikörper, die entweder gegen Aktivine oder GDFs gerichtet waren, um den jeweiligen Beitrag dieser Liganden zu den kardiopulmonalen In-vivo-Effekten von RAP-011 zu untersuchen, das Aktivine und GDFs mit hoher Affinität bindet und langsame Off-Raten aufweist, die für die Ligandensequestrierung vorteilhaft sind
23
. In einem präventiven SuHx-Rattenmodell (Abb.
3
wurden erhöhte hämodynamische Parameter wie der systolische Pulmonalarteriendruck (sPAP) und der mittlere Pulmonalarteriendruck (mPAP) effektiver durch eine duale Kombinationsbehandlung mit Antikörpern gegen Activin A/B und normalisiert GDF8/GDF11 als durch separate Antikörperbehandlungen (Abb.
3
C, D). Die duale Antikörperbehandlung normalisierte auch die RV-Hypertrophie effektiver als separate Antikörperbehandlungen (Abb.
3
E). Obwohl die Sequestrierung von Aktivinen oder GDFs in diesen Experimenten einen teilweisen Schutz vermittelte, implizieren unsere Ergebnisse, dass die Sequestrierung mehrerer Liganden des SMAD2/3-Wegs – wahrscheinlich Aktivine, GDF8 und GDF11 in Kombination – ein größeres Spektrum an therapeutischem Nutzen bietet und RAP-011 zugrunde liegt –induzierte Umkehrung kardiopulmonaler Beeinträchtigungen bei experimenteller PH.ActRIIA-Fc kehrt den Herzumbau bei schwerer experimenteller PAH umWir untersuchten die kardioprotektiven Wirkungen der RAP-011-Monotherapie weiter und verglichen sie mit Sildenafil im SuHxNx-Modell, das zu Beginn der therapeutischen Behandlung eine RV-Hypertrophie und einen beeinträchtigten Herzindex aufweist
28
(ergänzende Abb.
5
). Die therapeutische Behandlung mit RAP-011, beginnend 5 Wochen nach Krankheitsbeginn, verbesserte diese Parameter signifikant bis Woche 9 und führte zu einer größeren Verbesserung als die Behandlung mit Sildenafil (ergänzende Abb.
5
). Eine serielle Echokardiographie zeigte, dass die RAP-011-Therapie die RV-Dilatation, die Abflachung der Septumwand und die RV-Fraktionsflächenänderung (RVFAC) umkehrte, während Sildenafil dies nicht tat (Abb.
4
B, C). Wir bestätigten, dass die RAP-011-Therapie Anomalien der Pulmonalarterien-Beschleunigungszeit (PAAT) und der freien Wanddicke des RV (RVFWT) wirksamer linderte als Sildenafil (ergänzende Abb.
5
).Figur 4
Mehrere Liganden tragen zur Makrophagenaktivierung in vitro und zum kardiopulmonalen Umbau in einem SuHx-Rattenmodell von PH bei. ( A ) Expression von Markern der Makrophagenaktivierung in THP-1-Monozyten in vitro ohne Behandlung (Kontrolle) oder nach Behandlung mit Aktivin A (5 ng/ml), Aktivin B (50 ng/ml) oder GDF11 (5 ng/ml ). Analyse durch ungepaarten t-Test (* P < 0,05, ** P < 0,01 vs. Kontrolle). ( B ) Experimenteller Ansatz, der zum Testen der Wirkungen der Multi-Liganden-Hemmung in einem SuHx-Rattenmodell von PH verwendet wurde. Ratten wurden mit einer Einzeldosis von SU5416 (20 mg/kg, sc) behandelt und einer normobaren Hypoxie (13 % O2) ausgesetzt) und zweimal wöchentlich subkutan behandelt mit separaten Antikörpern gegen Aktivin A und Aktivin B (Anti-Act, jeweils 10 mg/kg), einem Antikörper mit doppelter Spezifität für GDF8 und GDF11 (Anti-GDF, 10 mg/kg), kombiniertem Anti – Act und Anti-GDF oder Vehikel (PBS) für 4 Wochen, beginnend einen Tag nach SU5416. ( C ) sPAP, ( D ) mPAP und ( E ) Fulton-Index. Die Daten sind Mittelwerte ± SEM (n = 5–9 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test (* P < 0,05, ** P < 0,01, **** P < 0,0001).
Wir verwendeten neutralisierende Antikörper, die entweder gegen Aktivine oder GDFs gerichtet waren, um den jeweiligen Beitrag dieser Liganden zu den kardiopulmonalen In-vivo-Effekten von RAP-011 zu untersuchen, das Aktivine und GDFs mit hoher Affinität bindet und langsame Off-Raten aufweist, die für die Ligandensequestrierung vorteilhaft sind
23
. In einem präventiven SuHx-Rattenmodell (Abb.
3
wurden erhöhte hämodynamische Parameter wie der systolische Pulmonalarteriendruck (sPAP) und der mittlere Pulmonalarteriendruck (mPAP) effektiver durch eine duale Kombinationsbehandlung mit Antikörpern gegen Activin A/B und normalisiert GDF8/GDF11 als durch separate Antikörperbehandlungen (Abb.
3
C, D). Die duale Antikörperbehandlung normalisierte auch die RV-Hypertrophie effektiver als separate Antikörperbehandlungen (Abb.
3
E). Obwohl die Sequestrierung von Aktivinen oder GDFs in diesen Experimenten einen teilweisen Schutz vermittelte, implizieren unsere Ergebnisse, dass die Sequestrierung mehrerer Liganden des SMAD2/3-Wegs – wahrscheinlich Aktivine, GDF8 und GDF11 in Kombination – ein größeres Spektrum an therapeutischem Nutzen bietet und RAP-011 zugrunde liegt –induzierte Umkehrung kardiopulmonaler Beeinträchtigungen bei experimenteller PH.ActRIIA-Fc kehrt den Herzumbau bei schwerer experimenteller PAH umWir untersuchten die kardioprotektiven Wirkungen der RAP-011-Monotherapie weiter und verglichen sie mit Sildenafil im SuHxNx-Modell, das zu Beginn der therapeutischen Behandlung eine RV-Hypertrophie und einen beeinträchtigten Herzindex aufweist
28
(ergänzende Abb.
5
). Die therapeutische Behandlung mit RAP-011, beginnend 5 Wochen nach Krankheitsbeginn, verbesserte diese Parameter signifikant bis Woche 9 und führte zu einer größeren Verbesserung als die Behandlung mit Sildenafil (ergänzende Abb.
5
). Eine serielle Echokardiographie zeigte, dass die RAP-011-Therapie die RV-Dilatation, die Abflachung der Septumwand und die RV-Fraktionsflächenänderung (RVFAC) umkehrte, während Sildenafil dies nicht tat (Abb.
4
B, C). Wir bestätigten, dass die RAP-011-Therapie Anomalien der Pulmonalarterien-Beschleunigungszeit (PAAT) und der freien Wanddicke des RV (RVFWT) wirksamer linderte als Sildenafil (ergänzende Abb.
5
).Figur 4
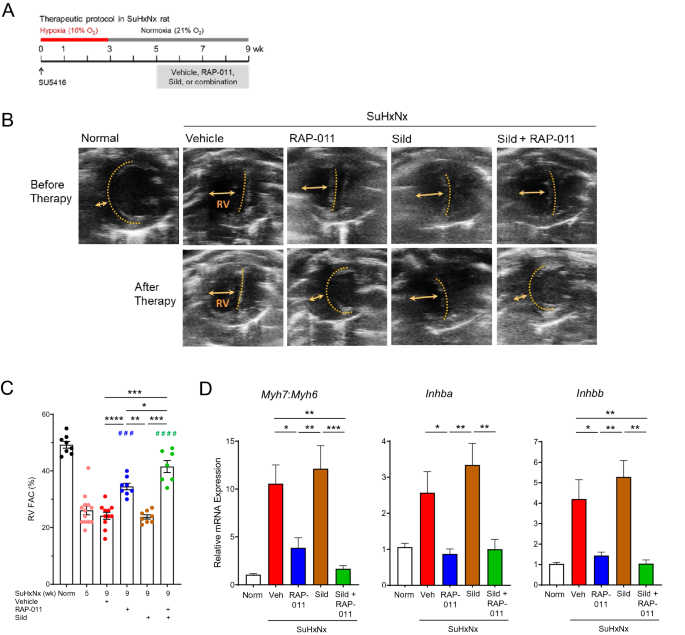 ActRIIA-Fc kehrt den kardialen Umbau und die Expression wichtiger kardialer Gene bei schwerer experimenteller PAH um. ( A ) Experimenteller Ansatz zur Bewertung der therapeutischen Wirkungen von RAP-011 im SuHxNx-Rattenmodell für schwere PAH. Siehe Abb.
1
für Einzelheiten. ( B ) Paare von repräsentativen echokardiographischen Bildern, die am Ende der Diastole von denselben SuHxNx-Ratten vor und nach der Therapie erhalten wurden. ( C ) RV-Teilbereichsänderung (RV FAC). Die Daten sind Mittelwerte ± SEM (n = 7–11 Ratten pro Gruppe). ( D ) Verhältnis der Myosin-Schwerketten-Isoform-Expression ( Myh7 : Myh6 ) und Spiegel von Inhba und Inhbbim RV von normalen oder SuHxNx-Ratten. Die Daten sind Mittelwerte ± SEM (n = 6–11 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-hoc-Test (*P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0,0001; ### P < 0,001 vs. Woche 5, # ### P < 0,0001 vs. Woche 5).
Wir untersuchten die Auswirkungen einer therapeutischen Behandlung mit RAP-011 auf ausgewählte molekulare Marker kardialer Dysfunktion im SuHxNx-Modell der schweren PAH. Herzumbau und Herzinsuffizienz sind mit einer erhöhten Activin-ActRIIA/B-Signalgebung und einer Expressionsverschiebung von der Isoform α der schweren Kette von Myosin zur Isoform β (erhöhtes Myh7 : Myh6- Verhältnis) verbunden
41
,
42
. Im Vergleich zu normalem Herzgewebe zeigte RV-Gewebe von mit Vehikel behandelten SuHxNx-Ratten in Woche 9 eine erhöhte Expression von β-Untereinheiten für Aktivin A ( Inhba ) und Aktivin B ( Inhbb ) und ein erhöhtes Verhältnis von Myh7 : Myh6- Expression (Fig.
4
D). In jedem Fall normalisierte die therapeutische Behandlung mit RAP-011 die Expression dieser Marker der Herzfunktionsstörung teilweise oder vollständig, während die Behandlung mit Vehikel oder Sildenafil dies nicht tat. Darüber hinaus verringerte die gleichzeitige Behandlung von SuHxNx-Ratten mit RAP-011 und Sildenafil die pSmad3-Spiegel im RV, und die RAP-011-Monotherapie reichte aus, um die pSmad1/5/8-Spiegel im RV zu erhöhen (ergänzende
6
). Zusammen erweitern diese Ergebnisse unsere früheren Erkenntnisse in einem SuHxNx-Modell und bestätigen, dass die Hemmung mehrerer Liganden der Activin-Klasse durch ActRIIA-Fc die anomale kardiale Genexpression umkehrt, den strukturellen Umbau des Herzens umkehrt und ein Ungleichgewicht in der RV-Smad-Signalübertragung bei schwerer experimenteller PAH teilweise korrigiert .ActRIIA-Fc ist wirksam, wenn es in Kombination mit einem Vasodilatator bei schwerer experimenteller PAH angewendet wirdAls Teil der oben beschriebenen Experimente mit SuHxNx-Ratten untersuchten wir, ob eine kombinierte Therapie mit RAP-011 und Sildenafil einen größeren therapeutischen Nutzen für eine etablierte Krankheit brachte als ihre jeweiligen Monotherapien. In zuvor nicht berichteten Vergleichen führte die kombinierte Therapie mit RAP-011 und Sildenafil zu einer signifikant größeren Verbesserung der kardialen Endpunkte als die Behandlung mit Sildenafil allein (Abb.
4
B–D; ergänzende Abb.
5
). Ein ähnliches Muster wurde für hämodynamische Defizite und Gefäßverschlüsse beobachtet (ergänzende Abb.
1
). Bei einigen Parametern schien die Kombinationstherapie wirksamer zu sein als die RAP-011-Monotherapie (Abb.
4
; ergänzende Abb.
1
,
5 ).
), obwohl der Großteil des Nutzens von RAP-011 bereitgestellt wurde. Diese Ergebnisse zeigen eine größere Wirksamkeit der ActRIIA-Fc-Monotherapie im Vergleich zur Sildenafil-Monotherapie sowie die Wirksamkeit von ActRIIA-Fc als Zusatztherapie in diesem Rattenmodell für schwere angioobliterative PAH, was mit der Wirksamkeit übereinstimmt, die bei PAH-Patienten beobachtet wurde, die eine Hintergrundtherapie erhielten
30
. Diese Ergebnisse untermauern die Ansicht, dass ActRIIA-Fc über Mechanismen wirkt, die sich weitgehend von denen aktueller PAH-Therapien unterscheiden.ActRIIA-Fc lindert den kardiopulmonalen Umbau und die Infiltration von Makrophagen in einem Modell für vererbbare PAH, die durch Bmpr2 -Haploinsuffizienz entstehtFunktionsverlust-Mutationen in BMPR2 wurden bei vererbbarer PAH identifiziert, und selbst idiopathische Formen von PAH sind entweder mit einer reduzierten BMPRII-Proteinexpression oder einer verminderten BMPRII-Signalgebung assoziiert
3
,
43
. Daher erzeugten wir Bmpr2-
haploinsuffiziente
Mäuse, wie von anderen berichtet
44
, und bewerteten die RAP-011-Aktivität in diesen mutierten Mäusen unter hypoxischen Bedingungen ( 5A ). Die Analyse der genomischen DNA bestätigte, dass Bmpr2 + /R899XMäuse besitzen eine heterozygote Nukleotidsubstitution an der erwarteten Position, und Immunoblotting bestätigte verringerte Mengen an BMPRII-Protein in Lungenlysaten, was mit einem verkürzten Proteinprodukt und/oder einem Nonsense-vermittelten mRNA-Abbau vereinbar ist (ergänzende
7
).Abbildung 5
ActRIIA-Fc kehrt den kardialen Umbau und die Expression wichtiger kardialer Gene bei schwerer experimenteller PAH um. ( A ) Experimenteller Ansatz zur Bewertung der therapeutischen Wirkungen von RAP-011 im SuHxNx-Rattenmodell für schwere PAH. Siehe Abb.
1
für Einzelheiten. ( B ) Paare von repräsentativen echokardiographischen Bildern, die am Ende der Diastole von denselben SuHxNx-Ratten vor und nach der Therapie erhalten wurden. ( C ) RV-Teilbereichsänderung (RV FAC). Die Daten sind Mittelwerte ± SEM (n = 7–11 Ratten pro Gruppe). ( D ) Verhältnis der Myosin-Schwerketten-Isoform-Expression ( Myh7 : Myh6 ) und Spiegel von Inhba und Inhbbim RV von normalen oder SuHxNx-Ratten. Die Daten sind Mittelwerte ± SEM (n = 6–11 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-hoc-Test (*P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0,0001; ### P < 0,001 vs. Woche 5, # ### P < 0,0001 vs. Woche 5).
Wir untersuchten die Auswirkungen einer therapeutischen Behandlung mit RAP-011 auf ausgewählte molekulare Marker kardialer Dysfunktion im SuHxNx-Modell der schweren PAH. Herzumbau und Herzinsuffizienz sind mit einer erhöhten Activin-ActRIIA/B-Signalgebung und einer Expressionsverschiebung von der Isoform α der schweren Kette von Myosin zur Isoform β (erhöhtes Myh7 : Myh6- Verhältnis) verbunden
41
,
42
. Im Vergleich zu normalem Herzgewebe zeigte RV-Gewebe von mit Vehikel behandelten SuHxNx-Ratten in Woche 9 eine erhöhte Expression von β-Untereinheiten für Aktivin A ( Inhba ) und Aktivin B ( Inhbb ) und ein erhöhtes Verhältnis von Myh7 : Myh6- Expression (Fig.
4
D). In jedem Fall normalisierte die therapeutische Behandlung mit RAP-011 die Expression dieser Marker der Herzfunktionsstörung teilweise oder vollständig, während die Behandlung mit Vehikel oder Sildenafil dies nicht tat. Darüber hinaus verringerte die gleichzeitige Behandlung von SuHxNx-Ratten mit RAP-011 und Sildenafil die pSmad3-Spiegel im RV, und die RAP-011-Monotherapie reichte aus, um die pSmad1/5/8-Spiegel im RV zu erhöhen (ergänzende
6
). Zusammen erweitern diese Ergebnisse unsere früheren Erkenntnisse in einem SuHxNx-Modell und bestätigen, dass die Hemmung mehrerer Liganden der Activin-Klasse durch ActRIIA-Fc die anomale kardiale Genexpression umkehrt, den strukturellen Umbau des Herzens umkehrt und ein Ungleichgewicht in der RV-Smad-Signalübertragung bei schwerer experimenteller PAH teilweise korrigiert .ActRIIA-Fc ist wirksam, wenn es in Kombination mit einem Vasodilatator bei schwerer experimenteller PAH angewendet wirdAls Teil der oben beschriebenen Experimente mit SuHxNx-Ratten untersuchten wir, ob eine kombinierte Therapie mit RAP-011 und Sildenafil einen größeren therapeutischen Nutzen für eine etablierte Krankheit brachte als ihre jeweiligen Monotherapien. In zuvor nicht berichteten Vergleichen führte die kombinierte Therapie mit RAP-011 und Sildenafil zu einer signifikant größeren Verbesserung der kardialen Endpunkte als die Behandlung mit Sildenafil allein (Abb.
4
B–D; ergänzende Abb.
5
). Ein ähnliches Muster wurde für hämodynamische Defizite und Gefäßverschlüsse beobachtet (ergänzende Abb.
1
). Bei einigen Parametern schien die Kombinationstherapie wirksamer zu sein als die RAP-011-Monotherapie (Abb.
4
; ergänzende Abb.
1
,
5 ).
), obwohl der Großteil des Nutzens von RAP-011 bereitgestellt wurde. Diese Ergebnisse zeigen eine größere Wirksamkeit der ActRIIA-Fc-Monotherapie im Vergleich zur Sildenafil-Monotherapie sowie die Wirksamkeit von ActRIIA-Fc als Zusatztherapie in diesem Rattenmodell für schwere angioobliterative PAH, was mit der Wirksamkeit übereinstimmt, die bei PAH-Patienten beobachtet wurde, die eine Hintergrundtherapie erhielten
30
. Diese Ergebnisse untermauern die Ansicht, dass ActRIIA-Fc über Mechanismen wirkt, die sich weitgehend von denen aktueller PAH-Therapien unterscheiden.ActRIIA-Fc lindert den kardiopulmonalen Umbau und die Infiltration von Makrophagen in einem Modell für vererbbare PAH, die durch Bmpr2 -Haploinsuffizienz entstehtFunktionsverlust-Mutationen in BMPR2 wurden bei vererbbarer PAH identifiziert, und selbst idiopathische Formen von PAH sind entweder mit einer reduzierten BMPRII-Proteinexpression oder einer verminderten BMPRII-Signalgebung assoziiert
3
,
43
. Daher erzeugten wir Bmpr2-
haploinsuffiziente
Mäuse, wie von anderen berichtet
44
, und bewerteten die RAP-011-Aktivität in diesen mutierten Mäusen unter hypoxischen Bedingungen ( 5A ). Die Analyse der genomischen DNA bestätigte, dass Bmpr2 + /R899XMäuse besitzen eine heterozygote Nukleotidsubstitution an der erwarteten Position, und Immunoblotting bestätigte verringerte Mengen an BMPRII-Protein in Lungenlysaten, was mit einem verkürzten Proteinprodukt und/oder einem Nonsense-vermittelten mRNA-Abbau vereinbar ist (ergänzende
7
).Abbildung 5
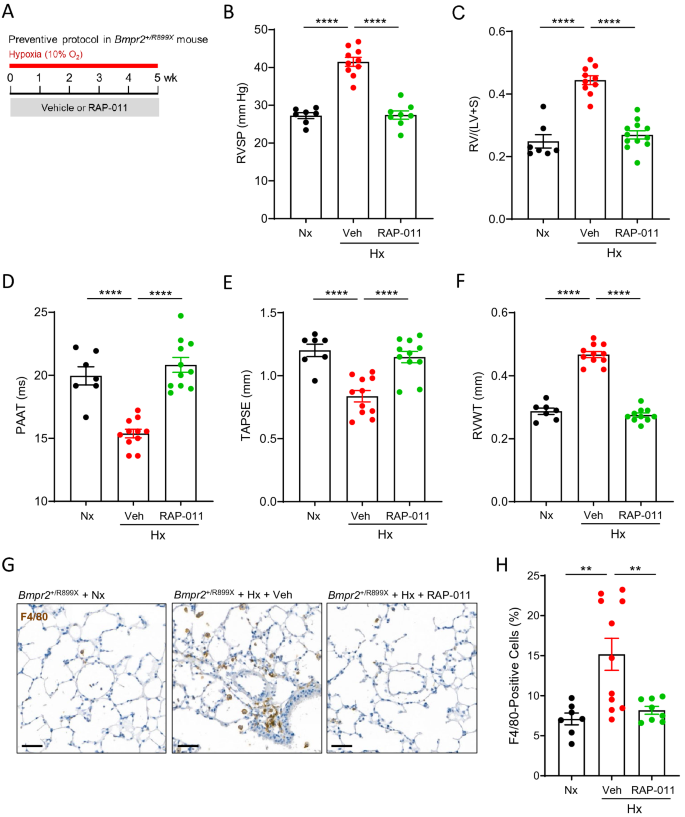 ActRIIA-Fc reduziert die Infiltration von Lungenmakrophagen und verhindert PH in einem Mausmodell der Bmpr2 -Haploinsuffizienz. ( A ) Experimenteller Ansatz zur Bewertung der kardiopulmonalen Wirkungen von RAP-011 bei Mäusen mit Bmpr2 - Haploinsuffizienz. Bmpr2 + /R899X- Mäuse wurden unter normoxischen Bedingungen (Nx) als Kontrollen gehalten oder einer normobaren Hypoxie (10 % O 2 ) ausgesetzt und zweimal wöchentlich entweder mit RAP-011 (10 mg/kg, sc) oder Vehikel (PBS) für behandelt 5 Wochen. ( B ) RVSP, ( C ) Fulton-Index, ( D ) PAAT, ( E ) TAPSE und ( F ) RVWT. ( G) Repräsentative Bilder von Lungenschnitten, die für den Makrophagenmarker F4/80 immungefärbt wurden. ( H ) Quantifizierung von F4/80-positiven Zellen in der Lunge basierend auf der Bewertung von 30 stark vergrößerten Feldern pro Maus. Maßstabsleiste, 50 µm. Die Daten sind Mittelwerte ± SEM (n = 7–10 pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test. * P < 0,05, ** P < 0,01, *** P < 0,001, **** P < 0,0001.
Die Exposition von Bmpr2 + /R899X- Mäusen gegenüber Hypoxie erhöhte RVSP, induzierte RV-Hypertrophie und erzeugte Anomalien bei PAAT, TAPSE und RVWT, während Bmpr2 + /R899X- Mäusen unter normoxischen Bedingungen diese kardiovaskulären Phänotypen fehlten (
5
B–F). Die vorbeugende Behandlung mit RAP-011 normalisierte jeden Endpunkt (Abb.
5
B–F). Darüber hinaus verursachte die Exposition von Bmpr2 + /R899X- Mäusen gegenüber Hypoxie für 5 Wochen eine Infiltration von Lungenmakrophagen (
5
G, H), wie durch Immunfärbung für den Makrophagenmarker F4/80 bestimmt. Ähnlich wie seine therapeutischen Wirkungen im SuHxNx-Rattenmodell verhinderte RAP-011 die Infiltration von Makrophagen in die Lungen von Bmpr2 + /R899X- Mäusen (
5
G, H). Im Gegensatz zu den Ergebnissen bei SuHxNx-Ratten (Abb. 2A
)
fanden wir jedoch keine Hinweise darauf, dass molekulare Entzündungsmarker in den Lungen von Bmpr2 + /R899X- Mäusen, die einer Hypoxie ausgesetzt waren, hochreguliert sind (ergänzende Abb.
8
), was darauf hindeutet, dass der entzündliche Phänotyp im Mausmodell weniger schwerwiegend ist. Zusammengenommen weisen diese Ergebnisse darauf hin, dass ein Mausmodell für vererbbare PAH wie das oben beschriebene SuHxNx-Rattenmodell für induzierte PAH durch ausgeprägte entzündliche Infiltrate gekennzeichnet ist und dass die Behandlung mit ActRIIA-Fc in jedem Fall mit einer unterdrückten Makrophageninfiltration und einer Wiederherstellung der kardiopulmonalen Funktion einhergeht Struktur und Funktion.Persistenz des ActRIIA-Fc-induzierten kardiopulmonalen Nutzens bei schwerer experimenteller PAHWir untersuchten, ob die kardiopulmonalen Vorteile der therapeutischen RAP-011-Behandlung bei schwerer experimenteller PAH nach Beendigung der Behandlung anhalten (Abb.
6
A). Bei unbehandelten SuHxNx-Ratten bestätigten wir, dass strukturelle und funktionelle Anomalien, die bis Woche 5 vorhanden waren, einschließlich veränderter RVSP, TPRI, RV-Hypertrophie, Herzindex, PAAT und TAPSE, bis Woche 13 weitgehend unverändert blieben (Abb.
6
B–G). Die therapeutische Behandlung mit RAP-011, beginnend in Woche 5, führte zu einer signifikanten Verbesserung dieser Parameter in Woche 9 (Abb.
6
B–G). Wichtig ist, dass bei SuHxNx-Ratten, die von Woche 5 bis 9 therapeutisch mit RAP-011 behandelt wurden, die Verbesserungen bei jedem dieser Endpunkte für 4 Wochen nach Beendigung der Behandlung bis Woche 13 anhielten (Abb.
6
BG). Zirkulierende RAP-011-Spiegel waren 2 Wochen nach Absetzen der Behandlung nicht nachweisbar. Diese Ergebnisse weisen darauf hin, dass die gleichzeitige Hemmung von Liganden der Activin-Klasse und die Blockade von Entzündungsprozessen durch ActRIIA-Fc zu einer anhaltenden Umkehrung des kardiopulmonalen strukturellen Umbaus bei schwerer experimenteller PAH führt, selbst in der offensichtlichen Abwesenheit aktiv zirkulierender therapeutischer Moleküle.Abbildung 6
ActRIIA-Fc reduziert die Infiltration von Lungenmakrophagen und verhindert PH in einem Mausmodell der Bmpr2 -Haploinsuffizienz. ( A ) Experimenteller Ansatz zur Bewertung der kardiopulmonalen Wirkungen von RAP-011 bei Mäusen mit Bmpr2 - Haploinsuffizienz. Bmpr2 + /R899X- Mäuse wurden unter normoxischen Bedingungen (Nx) als Kontrollen gehalten oder einer normobaren Hypoxie (10 % O 2 ) ausgesetzt und zweimal wöchentlich entweder mit RAP-011 (10 mg/kg, sc) oder Vehikel (PBS) für behandelt 5 Wochen. ( B ) RVSP, ( C ) Fulton-Index, ( D ) PAAT, ( E ) TAPSE und ( F ) RVWT. ( G) Repräsentative Bilder von Lungenschnitten, die für den Makrophagenmarker F4/80 immungefärbt wurden. ( H ) Quantifizierung von F4/80-positiven Zellen in der Lunge basierend auf der Bewertung von 30 stark vergrößerten Feldern pro Maus. Maßstabsleiste, 50 µm. Die Daten sind Mittelwerte ± SEM (n = 7–10 pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test. * P < 0,05, ** P < 0,01, *** P < 0,001, **** P < 0,0001.
Die Exposition von Bmpr2 + /R899X- Mäusen gegenüber Hypoxie erhöhte RVSP, induzierte RV-Hypertrophie und erzeugte Anomalien bei PAAT, TAPSE und RVWT, während Bmpr2 + /R899X- Mäusen unter normoxischen Bedingungen diese kardiovaskulären Phänotypen fehlten (
5
B–F). Die vorbeugende Behandlung mit RAP-011 normalisierte jeden Endpunkt (Abb.
5
B–F). Darüber hinaus verursachte die Exposition von Bmpr2 + /R899X- Mäusen gegenüber Hypoxie für 5 Wochen eine Infiltration von Lungenmakrophagen (
5
G, H), wie durch Immunfärbung für den Makrophagenmarker F4/80 bestimmt. Ähnlich wie seine therapeutischen Wirkungen im SuHxNx-Rattenmodell verhinderte RAP-011 die Infiltration von Makrophagen in die Lungen von Bmpr2 + /R899X- Mäusen (
5
G, H). Im Gegensatz zu den Ergebnissen bei SuHxNx-Ratten (Abb. 2A
)
fanden wir jedoch keine Hinweise darauf, dass molekulare Entzündungsmarker in den Lungen von Bmpr2 + /R899X- Mäusen, die einer Hypoxie ausgesetzt waren, hochreguliert sind (ergänzende Abb.
8
), was darauf hindeutet, dass der entzündliche Phänotyp im Mausmodell weniger schwerwiegend ist. Zusammengenommen weisen diese Ergebnisse darauf hin, dass ein Mausmodell für vererbbare PAH wie das oben beschriebene SuHxNx-Rattenmodell für induzierte PAH durch ausgeprägte entzündliche Infiltrate gekennzeichnet ist und dass die Behandlung mit ActRIIA-Fc in jedem Fall mit einer unterdrückten Makrophageninfiltration und einer Wiederherstellung der kardiopulmonalen Funktion einhergeht Struktur und Funktion.Persistenz des ActRIIA-Fc-induzierten kardiopulmonalen Nutzens bei schwerer experimenteller PAHWir untersuchten, ob die kardiopulmonalen Vorteile der therapeutischen RAP-011-Behandlung bei schwerer experimenteller PAH nach Beendigung der Behandlung anhalten (Abb.
6
A). Bei unbehandelten SuHxNx-Ratten bestätigten wir, dass strukturelle und funktionelle Anomalien, die bis Woche 5 vorhanden waren, einschließlich veränderter RVSP, TPRI, RV-Hypertrophie, Herzindex, PAAT und TAPSE, bis Woche 13 weitgehend unverändert blieben (Abb.
6
B–G). Die therapeutische Behandlung mit RAP-011, beginnend in Woche 5, führte zu einer signifikanten Verbesserung dieser Parameter in Woche 9 (Abb.
6
B–G). Wichtig ist, dass bei SuHxNx-Ratten, die von Woche 5 bis 9 therapeutisch mit RAP-011 behandelt wurden, die Verbesserungen bei jedem dieser Endpunkte für 4 Wochen nach Beendigung der Behandlung bis Woche 13 anhielten (Abb.
6
BG). Zirkulierende RAP-011-Spiegel waren 2 Wochen nach Absetzen der Behandlung nicht nachweisbar. Diese Ergebnisse weisen darauf hin, dass die gleichzeitige Hemmung von Liganden der Activin-Klasse und die Blockade von Entzündungsprozessen durch ActRIIA-Fc zu einer anhaltenden Umkehrung des kardiopulmonalen strukturellen Umbaus bei schwerer experimenteller PAH führt, selbst in der offensichtlichen Abwesenheit aktiv zirkulierender therapeutischer Moleküle.Abbildung 6
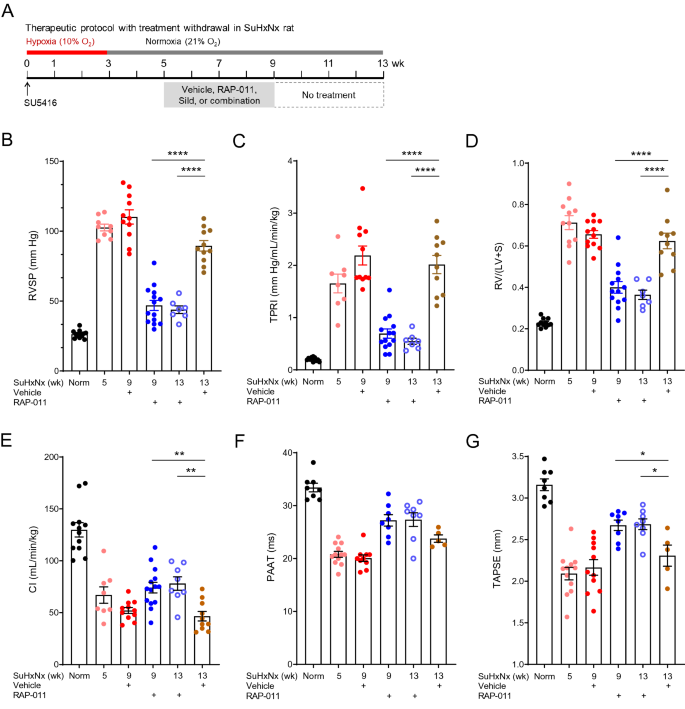 Die krankheitsaufhebenden Wirkungen von ActRIIA-Fc bei schwerer experimenteller PAH bleiben nach Beendigung der Behandlung bestehen. ( A ) Experimenteller Ansatz zur Bewertung der Persistenz der therapeutischen Wirkungen von RAP-011 in einem SuHxNx-Rattenmodell für schwere PAH. Ratten wurden am Tag 0 mit einer Einzeldosis von SU5416 (20 mg/kg, sc) behandelt und 3 Wochen einer normobaren Hypoxie (10 % O 2 ) ausgesetzt, gefolgt von 10 Wochen Normoxie, um ein Fortschreiten der Krankheit zu ermöglichen. Ratten wurden zusätzlich zweimal wöchentlich mit RAP-011 (2,5 mg/kg, sc) oder Vehikel (PBS) von Woche 5 bis Woche 9 nach SU5416 behandelt, zu diesem Zeitpunkt wurde die Behandlung für die verbleibenden 4 Wochen unterbrochen. ( B ) RVSP, ( C ) TPRI, ( D ) Fulton-Index, ( E ) Herzindex (CI), (F ) PAAT und ( G ) TAPSE. Die Daten sind Mittelwerte ± SEM (n = 7–13 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test (* P < 0,05, ** P < 0,01, **** P < 0,0001).
DiskussionDie komplexe, multifaktorielle Ätiologie der PAH stellt eine gewaltige Herausforderung für die Entwicklung krankheitsmodifizierender Therapien dar. Die geringe Penetranz mutierter Allele wie BMPR2
3
,
4
weist darauf hin, dass zusätzlich zum BMPRII-Mangel Faktoren erforderlich sind, um PAH in den meisten Fällen zu induzieren, und unterstreicht die Bedeutung therapeutischer Ansätze, die auf mehrere Krankheitsmediatoren in Kombination abzielen. Wir haben zuvor eine unerwartet herausragende Rolle von Liganden der Activin-Klasse als Treiber von Lungengefäßerkrankungen identifiziert und ActRIIA-Fc als potenziellen therapeutischen Ansatz zur Wiederherstellung des Gleichgewichts zwischen der pulmonalvaskulären SMAD1/5/8- und SMAD2/3-Signalübertragung bei PAH etabliert
28
. Hier identifizieren wir Liganden der Activin-Klasse als Schlüsselmediatoren von Entzündungs- und Immunantworten – entweder direkt oder indirekt – bei schwerer experimenteller PAH und weisen auf wichtige regulatorische Wirkungen dieser Liganden auf die Aktivierung von Makrophagen und die perivaskuläre Infiltration in erkranktem Lungengewebe hin. Diese Ergebnisse sind die ersten, die eine durch Aktivin getriebene Entzündung bei der Umgestaltung der Lungengefäße bei PAH implizieren und das Spektrum bekannter pathologischer Wirkungen für diese wichtigen Liganden der TGF-β-Superfamilie erweitern.Unsere Ergebnisse zeigen, dass entzündliche Gensignaturen und perivaskuläre Infiltrate von Makrophagen bei schwerer experimenteller PAH durch die therapeutische Behandlung mit ActRIIA-Fc normalisiert wurden, und die von uns beobachtete Übereinstimmung zwischen abweichenden Genprofilen in diesem Nagetiermodell und bei PAH-Patienten unterstützt nachdrücklich die Übertragbarkeit dieser Ergebnisse auf menschliche PAH. Wie durch die Hauptkomponentenanalyse bestätigt wurde, kehrte die therapeutische Behandlung mit ActRIIA-Fc diesen entzündlichen pathologischen Phänotyp in einem Ausmaß um, das das eines Standard-Vasodilatators weit übertraf. ActRIIA-Fc verhinderte in ähnlicher Weise das Eindringen von Makrophagen in das Lungengewebe und übte in zwei anderen Modellen, insbesondere in einem Mausmodell von vererbbarer PAH, die aus Bmpr2 hervorgeht , vorteilhafte kardiopulmonale Wirkungen ausHaploinsuffizienz. Darüber hinaus unterstützen unsere Ergebnisse einen Krankheitsmechanismus, bei dem Aktivine und GDFs mit überlappenden Aktivitätsprofilen konzertiert wirken, um eine Lungenentzündung und einen kardiopulmonalen Umbau zu fördern. Während also die Blockade von entweder Aktivinen oder GDFs mit ihren jeweiligen Antikörpern strukturelle und funktionelle Verbesserungen hervorrief, verlieh ihre kombinierte Neutralisierung einen zusätzlichen Vorteil. Schließlich wurden die Anti-Remodeling-Effekte der ActRIIA-Fc-Behandlung bei schwerer experimenteller PAH durch die gleichzeitige Vasodilatatortherapie nicht verringert, was auf das Potenzial dieses Wirkstoffs als wirksame Zusatztherapie sowie als Monotherapie hinweist. Die vorteilhaften Wirkungen hielten mindestens einen Monat nach Beendigung der Behandlung an, was darauf hindeutet, dass ActRIIA-Fc im Gegensatz zu Standard-Vasodilatatoren krankheitsmodifizierend wirken könnte.Es besteht ein wachsender Konsens darüber, dass frühe und anhaltende Entzündungen und veränderte Immunantworten der PAH-Pathophysiologie zugrunde liegen. Es wurde vorgeschlagen, dass der fortgeschrittene Gefäßumbau durch Ansätze reversibel sein könnte, die sich mit spezifischen Entzündungs- und Immunprozessen befassen
2
. In Übereinstimmung mit der normalen entzündungshemmenden Rolle von BMPRII in pulmonalen Endothelzellen
11
,
12
,
13
wurde eine Entzündung als wahrscheinlicher zweiter Treffer impliziert, der erforderlich ist, um eine schwere vaskuläre Pathologie im Zusammenhang mit einer reduzierten BMPRII-Signalübertragung zu induzieren
14
. In einer Studie fanden Tian und Mitarbeiter heraus, dass ein akuter entzündlicher Insult eine mesenchymale Transdifferenzierung durch pulmonale Endothelzellen (EndMT) durch Aktivierung der kanonischen SMAD2/3-Signalgebung verursachte, ein Effekt, der in vitro durch Knockdown von Tgfbr1 (ALK5) oder Smad3 reversibel war und reversibel in Bmpr2 -Mutantenratten durch einen niedermolekularen Inhibitor von ALK5. Ihre Ergebnisse deuten ferner darauf hin, dass SMAD1/5/8- und SMAD2/3-Signalweg-Wechselwirkungen ein wichtiger Punkt für die Konvergenz früher pathogener Faktoren bei PAH sind
10
; Es ist jedoch bemerkenswert, dass Interventionen, die auf ALK5 oder SMAD3 abzielen, möglicherweise auch die Signalübertragung durch Aktivine, GDF8 und GDF11, hemmen könnten, die nachgeschaltete Effektoren mit TGF-β teilen.Unsere Ergebnisse identifizieren perivaskuläre Makrophagen als einen wichtigen Zelltyp, durch den ActRIIA-Fc den Lungengefäßumbau bei schwerer experimenteller PAH umkehrt. Obwohl ihre Rolle weiter untersucht werden muss, sind Monozyten-Makrophagen stark an PAH beteiligt
45
,
46
,
47
,
48
,
49
, die möglicherweise sowohl die Initiierung als auch die Auflösung von Lungenentzündungen orchestrieren. Insbesondere Activin A ist umfassend an der Aktivierung von Makrophagen, Entzündungen und Fibrose beteiligt
33
,
50
,
51
,
52
. Bei PAH-Patienten produzieren alveoläre Makrophagen Aktivin A, und erhöhte Konzentrationen von zirkulierendem Aktivin A sind prädiktiv für die Sterblichkeit des Patienten
25
. Unsere Ergebnisse zeigen, dass Liganden der Activin-Klasse unterschiedliche, aber überlappende Wirkungen auf Monozyten-Makrophagen ausüben können, wobei eine gemeinsame Wirkung von Activin A und GDF11 die erhöhte Expression von Ccl2 ist, das für ein wichtiges Chemokin kodiert, das die Monozyten-Makrophagen-Chemotaxis und die Endothelpermeabilität fördert
53
. Im Gegensatz zur CCL2-fördernden Wirkung der SMAD2/3-Weg-Aktivatoren hemmen BMP9 und BMP10 die Freisetzung von CCL2 durch Lungenendothelzellen, um die Gefäßruhe zu fördern
54
. Die reziproke Regulation von CCL2 durch Liganden der Activin-Klasse und BMPs legt nahe, dass CCL2 ein Schlüsselmediator sein könnte, durch den Liganden der Activin-Klasse die vaskuläre Ruhe stören und den pathologischen Umbau fördern – ein elegantes Beispiel für eine bidirektionale homöostatische Regulation durch SMAD2/3 und SMAD1/5/8 Wege. Darüber hinaus unterstreicht die gemeinsame Regulation von CCL2 durch Activin A und GDF11 die Notwendigkeit, mehrere Liganden gleichzeitig anzugreifen, um ein robustes therapeutisches Ergebnis zu erzielen.Ein prominentes proinflammatorisches Zytokin, das in unserer Studie identifiziert wurde, ist IL-6, dessen Genexpression bei schwerer experimenteller PAH erhöht ist und durch die ActRIIA-Fc-Therapie normalisiert wird. Darüber hinaus fanden wir heraus, dass Aktivin A, Aktivin B und GDF11 jeweils die Il6- Expressionsniveaus als eine Komponente ihrer proinflammatorischen Wirkungen auf Monozyten-Makrophagen in vitro erhöhen. IL-6 vermittelt die Aktivierung von Lungenmakrophagen durch Adventitia-Fibroblasten, ist an menschlicher PAH beteiligt und verursacht bei Überexpression die spontane Entwicklung von PH bei Mäusen
47
,
55
,
56
,
57
,
58
,
59
. Darüber hinaus zeigen BMPRII- und IL-6-assoziierte Signalwege eine reziproke Regulation in pulmonalen glatten Muskelzellen
60
, was noch eine weitere mögliche Verbindung zwischen BMPRII-Signalisierung und Entzündung in der PAH-Pathogenese bereitstellt. Die umfangreichen Beweise für eine pathogene Rolle von IL-6 legten nahe, dass dieses Zytokin gezielt eingesetzt werden könnte, um Entzündungen zu reduzieren und dadurch andere Komponenten der PAH-Krankheit abzuschwächen. Eine klinische Studie an Patienten mit PAH konnte jedoch keine robusten hämodynamischen Vorteile von Tocilizumab, einem gegen den IL-6-Rezeptor gerichteten monoklonalen Antikörper, nachweisen
61
. Obwohl Folgestudien mit größeren Patientenpopulationen erforderlich sein werden, deuten diese Ergebnisse darauf hin, dass die alleinige Bekämpfung von Entzündungen für die Behandlung von PAH möglicherweise nicht ausreicht, und unterstreichen die komplexen, multifaktoriellen Mechanismen des Fortschreitens der PAH-Krankheit.Es ist besonders bemerkenswert, dass die ActRIIA-Fc-Behandlung bei schwerer experimenteller PAH die erhöhte pulmonale Expression von Grem1 umkehrt , das für einen endogenen BMP-Antagonisten (Gremlin-1) kodiert, der als wichtiger Förderer des Gefäßumbaus bei PAH angesehen wird
38
. Hypoxie stimuliert die Gremlin-Sekretion durch pulmonale mikrovaskuläre Endothelzellen, und Grem1 -Haploinsuffizienz reduziert den pulmonalen Gefäßumbau bei Mäusen, die chronischer Hypoxie ausgesetzt waren
38
. Andere Zelltypen, einschließlich arterieller glatter Muskelzellen und Makrophagen, könnten ebenfalls Gremlin-1-Quellen sein, und ersterer Typ zeigt eine erhöhte Gremlin-1-Expression als Reaktion auf mechanische Dehnung in vitro
62
,
63
. Gremlin-1 ist am besten für seine proproliferativen Wirkungen bekannt und wurde auch mit Entzündungen in der Niere und der Lunge in Verbindung gebracht, und zwar durch Auswirkungen auf die Notch-Signalübertragung und die Migration von Makrophagen
62
,
64
. Gremlin-1 spielt eine wichtige Rolle bei PAH im Zusammenhang mit angeborenen Herzfehlern (systemisch-pulmonale Shunts), die typischerweise nicht aus einer BMPR2 - Mutation resultieren; in diesem Fall könnte Gremlin-1 helfen, die verringerte Aktivität des BMPRII-SMAD1/5/8-Wegs in Gegenwart von intaktem BMPR2 zu erklären
63
. Wichtig ist, dass die therapeutische Immunneutralisierung von Gremlin-1 den pulmonalen Gefäßumbau bei experimenteller PAH reduziert
65
. Also Umkehrung von Grem1Überexpression könnte ein Schlüsselmechanismus sein, durch den ActRIIA-Fc die SMAD1/5/8-Signalisierung mit der SMAD2/3-Signalisierung in den Lungengefäßen wieder ins Gleichgewicht bringt
28
. Da Gremlin-1 als Mediator der Hemmung des BMPRII-Signalwegs durch Endothelin
66
fungiert, könnte die Gremlin-1-Modulation auch ein potenzieller Punkt der mechanistischen Konvergenz zwischen den therapeutischen Wirkungen von ActRIIA-Fc und denen von Endothelin-Rezeptor-Antagonisten bei Patienten mit PAH sein.Der vaskuläre Umbau bei PAH wird allgemein so verstanden, dass er aus physiologischen zellulären Reaktionen auf Stress oder Verletzungen entsteht, die schließlich dysreguliert und anhaltend werden. Entzündung, Hypoxie oder biomechanischer Stress bei Personen mit eingeschränkter Aktivität des BMPRII-Signalwegs können eine pathologische Remodellierung der extrazellulären Matrix, abnormale Zellproliferation und möglicherweise EndMT fördern
67
. Jüngste Ergebnisse deuten darauf hin, dass BMPRII eine schützende Rolle bei der Homöostase von Endothelzellen spielt, wobei der Verlust von BMPRII EndMT begünstigt und die Zellen in einen vorbereiteten biomechanischen Zustand treibt, in dem Änderungen der Steifheit oder Scherspannung einen zweiten Schlag liefern und einen selbsterhaltenden Zyklus von überschüssigem TGF einleiten -β-Signalisierung
10
. Die chronische TGF-β1-Signalgebung
Die krankheitsaufhebenden Wirkungen von ActRIIA-Fc bei schwerer experimenteller PAH bleiben nach Beendigung der Behandlung bestehen. ( A ) Experimenteller Ansatz zur Bewertung der Persistenz der therapeutischen Wirkungen von RAP-011 in einem SuHxNx-Rattenmodell für schwere PAH. Ratten wurden am Tag 0 mit einer Einzeldosis von SU5416 (20 mg/kg, sc) behandelt und 3 Wochen einer normobaren Hypoxie (10 % O 2 ) ausgesetzt, gefolgt von 10 Wochen Normoxie, um ein Fortschreiten der Krankheit zu ermöglichen. Ratten wurden zusätzlich zweimal wöchentlich mit RAP-011 (2,5 mg/kg, sc) oder Vehikel (PBS) von Woche 5 bis Woche 9 nach SU5416 behandelt, zu diesem Zeitpunkt wurde die Behandlung für die verbleibenden 4 Wochen unterbrochen. ( B ) RVSP, ( C ) TPRI, ( D ) Fulton-Index, ( E ) Herzindex (CI), (F ) PAAT und ( G ) TAPSE. Die Daten sind Mittelwerte ± SEM (n = 7–13 Ratten pro Gruppe). Analyse durch einfache ANOVA und Tukey-Post-Hoc-Test (* P < 0,05, ** P < 0,01, **** P < 0,0001).
DiskussionDie komplexe, multifaktorielle Ätiologie der PAH stellt eine gewaltige Herausforderung für die Entwicklung krankheitsmodifizierender Therapien dar. Die geringe Penetranz mutierter Allele wie BMPR2
3
,
4
weist darauf hin, dass zusätzlich zum BMPRII-Mangel Faktoren erforderlich sind, um PAH in den meisten Fällen zu induzieren, und unterstreicht die Bedeutung therapeutischer Ansätze, die auf mehrere Krankheitsmediatoren in Kombination abzielen. Wir haben zuvor eine unerwartet herausragende Rolle von Liganden der Activin-Klasse als Treiber von Lungengefäßerkrankungen identifiziert und ActRIIA-Fc als potenziellen therapeutischen Ansatz zur Wiederherstellung des Gleichgewichts zwischen der pulmonalvaskulären SMAD1/5/8- und SMAD2/3-Signalübertragung bei PAH etabliert
28
. Hier identifizieren wir Liganden der Activin-Klasse als Schlüsselmediatoren von Entzündungs- und Immunantworten – entweder direkt oder indirekt – bei schwerer experimenteller PAH und weisen auf wichtige regulatorische Wirkungen dieser Liganden auf die Aktivierung von Makrophagen und die perivaskuläre Infiltration in erkranktem Lungengewebe hin. Diese Ergebnisse sind die ersten, die eine durch Aktivin getriebene Entzündung bei der Umgestaltung der Lungengefäße bei PAH implizieren und das Spektrum bekannter pathologischer Wirkungen für diese wichtigen Liganden der TGF-β-Superfamilie erweitern.Unsere Ergebnisse zeigen, dass entzündliche Gensignaturen und perivaskuläre Infiltrate von Makrophagen bei schwerer experimenteller PAH durch die therapeutische Behandlung mit ActRIIA-Fc normalisiert wurden, und die von uns beobachtete Übereinstimmung zwischen abweichenden Genprofilen in diesem Nagetiermodell und bei PAH-Patienten unterstützt nachdrücklich die Übertragbarkeit dieser Ergebnisse auf menschliche PAH. Wie durch die Hauptkomponentenanalyse bestätigt wurde, kehrte die therapeutische Behandlung mit ActRIIA-Fc diesen entzündlichen pathologischen Phänotyp in einem Ausmaß um, das das eines Standard-Vasodilatators weit übertraf. ActRIIA-Fc verhinderte in ähnlicher Weise das Eindringen von Makrophagen in das Lungengewebe und übte in zwei anderen Modellen, insbesondere in einem Mausmodell von vererbbarer PAH, die aus Bmpr2 hervorgeht , vorteilhafte kardiopulmonale Wirkungen ausHaploinsuffizienz. Darüber hinaus unterstützen unsere Ergebnisse einen Krankheitsmechanismus, bei dem Aktivine und GDFs mit überlappenden Aktivitätsprofilen konzertiert wirken, um eine Lungenentzündung und einen kardiopulmonalen Umbau zu fördern. Während also die Blockade von entweder Aktivinen oder GDFs mit ihren jeweiligen Antikörpern strukturelle und funktionelle Verbesserungen hervorrief, verlieh ihre kombinierte Neutralisierung einen zusätzlichen Vorteil. Schließlich wurden die Anti-Remodeling-Effekte der ActRIIA-Fc-Behandlung bei schwerer experimenteller PAH durch die gleichzeitige Vasodilatatortherapie nicht verringert, was auf das Potenzial dieses Wirkstoffs als wirksame Zusatztherapie sowie als Monotherapie hinweist. Die vorteilhaften Wirkungen hielten mindestens einen Monat nach Beendigung der Behandlung an, was darauf hindeutet, dass ActRIIA-Fc im Gegensatz zu Standard-Vasodilatatoren krankheitsmodifizierend wirken könnte.Es besteht ein wachsender Konsens darüber, dass frühe und anhaltende Entzündungen und veränderte Immunantworten der PAH-Pathophysiologie zugrunde liegen. Es wurde vorgeschlagen, dass der fortgeschrittene Gefäßumbau durch Ansätze reversibel sein könnte, die sich mit spezifischen Entzündungs- und Immunprozessen befassen
2
. In Übereinstimmung mit der normalen entzündungshemmenden Rolle von BMPRII in pulmonalen Endothelzellen
11
,
12
,
13
wurde eine Entzündung als wahrscheinlicher zweiter Treffer impliziert, der erforderlich ist, um eine schwere vaskuläre Pathologie im Zusammenhang mit einer reduzierten BMPRII-Signalübertragung zu induzieren
14
. In einer Studie fanden Tian und Mitarbeiter heraus, dass ein akuter entzündlicher Insult eine mesenchymale Transdifferenzierung durch pulmonale Endothelzellen (EndMT) durch Aktivierung der kanonischen SMAD2/3-Signalgebung verursachte, ein Effekt, der in vitro durch Knockdown von Tgfbr1 (ALK5) oder Smad3 reversibel war und reversibel in Bmpr2 -Mutantenratten durch einen niedermolekularen Inhibitor von ALK5. Ihre Ergebnisse deuten ferner darauf hin, dass SMAD1/5/8- und SMAD2/3-Signalweg-Wechselwirkungen ein wichtiger Punkt für die Konvergenz früher pathogener Faktoren bei PAH sind
10
; Es ist jedoch bemerkenswert, dass Interventionen, die auf ALK5 oder SMAD3 abzielen, möglicherweise auch die Signalübertragung durch Aktivine, GDF8 und GDF11, hemmen könnten, die nachgeschaltete Effektoren mit TGF-β teilen.Unsere Ergebnisse identifizieren perivaskuläre Makrophagen als einen wichtigen Zelltyp, durch den ActRIIA-Fc den Lungengefäßumbau bei schwerer experimenteller PAH umkehrt. Obwohl ihre Rolle weiter untersucht werden muss, sind Monozyten-Makrophagen stark an PAH beteiligt
45
,
46
,
47
,
48
,
49
, die möglicherweise sowohl die Initiierung als auch die Auflösung von Lungenentzündungen orchestrieren. Insbesondere Activin A ist umfassend an der Aktivierung von Makrophagen, Entzündungen und Fibrose beteiligt
33
,
50
,
51
,
52
. Bei PAH-Patienten produzieren alveoläre Makrophagen Aktivin A, und erhöhte Konzentrationen von zirkulierendem Aktivin A sind prädiktiv für die Sterblichkeit des Patienten
25
. Unsere Ergebnisse zeigen, dass Liganden der Activin-Klasse unterschiedliche, aber überlappende Wirkungen auf Monozyten-Makrophagen ausüben können, wobei eine gemeinsame Wirkung von Activin A und GDF11 die erhöhte Expression von Ccl2 ist, das für ein wichtiges Chemokin kodiert, das die Monozyten-Makrophagen-Chemotaxis und die Endothelpermeabilität fördert
53
. Im Gegensatz zur CCL2-fördernden Wirkung der SMAD2/3-Weg-Aktivatoren hemmen BMP9 und BMP10 die Freisetzung von CCL2 durch Lungenendothelzellen, um die Gefäßruhe zu fördern
54
. Die reziproke Regulation von CCL2 durch Liganden der Activin-Klasse und BMPs legt nahe, dass CCL2 ein Schlüsselmediator sein könnte, durch den Liganden der Activin-Klasse die vaskuläre Ruhe stören und den pathologischen Umbau fördern – ein elegantes Beispiel für eine bidirektionale homöostatische Regulation durch SMAD2/3 und SMAD1/5/8 Wege. Darüber hinaus unterstreicht die gemeinsame Regulation von CCL2 durch Activin A und GDF11 die Notwendigkeit, mehrere Liganden gleichzeitig anzugreifen, um ein robustes therapeutisches Ergebnis zu erzielen.Ein prominentes proinflammatorisches Zytokin, das in unserer Studie identifiziert wurde, ist IL-6, dessen Genexpression bei schwerer experimenteller PAH erhöht ist und durch die ActRIIA-Fc-Therapie normalisiert wird. Darüber hinaus fanden wir heraus, dass Aktivin A, Aktivin B und GDF11 jeweils die Il6- Expressionsniveaus als eine Komponente ihrer proinflammatorischen Wirkungen auf Monozyten-Makrophagen in vitro erhöhen. IL-6 vermittelt die Aktivierung von Lungenmakrophagen durch Adventitia-Fibroblasten, ist an menschlicher PAH beteiligt und verursacht bei Überexpression die spontane Entwicklung von PH bei Mäusen
47
,
55
,
56
,
57
,
58
,
59
. Darüber hinaus zeigen BMPRII- und IL-6-assoziierte Signalwege eine reziproke Regulation in pulmonalen glatten Muskelzellen
60
, was noch eine weitere mögliche Verbindung zwischen BMPRII-Signalisierung und Entzündung in der PAH-Pathogenese bereitstellt. Die umfangreichen Beweise für eine pathogene Rolle von IL-6 legten nahe, dass dieses Zytokin gezielt eingesetzt werden könnte, um Entzündungen zu reduzieren und dadurch andere Komponenten der PAH-Krankheit abzuschwächen. Eine klinische Studie an Patienten mit PAH konnte jedoch keine robusten hämodynamischen Vorteile von Tocilizumab, einem gegen den IL-6-Rezeptor gerichteten monoklonalen Antikörper, nachweisen
61
. Obwohl Folgestudien mit größeren Patientenpopulationen erforderlich sein werden, deuten diese Ergebnisse darauf hin, dass die alleinige Bekämpfung von Entzündungen für die Behandlung von PAH möglicherweise nicht ausreicht, und unterstreichen die komplexen, multifaktoriellen Mechanismen des Fortschreitens der PAH-Krankheit.Es ist besonders bemerkenswert, dass die ActRIIA-Fc-Behandlung bei schwerer experimenteller PAH die erhöhte pulmonale Expression von Grem1 umkehrt , das für einen endogenen BMP-Antagonisten (Gremlin-1) kodiert, der als wichtiger Förderer des Gefäßumbaus bei PAH angesehen wird
38
. Hypoxie stimuliert die Gremlin-Sekretion durch pulmonale mikrovaskuläre Endothelzellen, und Grem1 -Haploinsuffizienz reduziert den pulmonalen Gefäßumbau bei Mäusen, die chronischer Hypoxie ausgesetzt waren
38
. Andere Zelltypen, einschließlich arterieller glatter Muskelzellen und Makrophagen, könnten ebenfalls Gremlin-1-Quellen sein, und ersterer Typ zeigt eine erhöhte Gremlin-1-Expression als Reaktion auf mechanische Dehnung in vitro
62
,
63
. Gremlin-1 ist am besten für seine proproliferativen Wirkungen bekannt und wurde auch mit Entzündungen in der Niere und der Lunge in Verbindung gebracht, und zwar durch Auswirkungen auf die Notch-Signalübertragung und die Migration von Makrophagen
62
,
64
. Gremlin-1 spielt eine wichtige Rolle bei PAH im Zusammenhang mit angeborenen Herzfehlern (systemisch-pulmonale Shunts), die typischerweise nicht aus einer BMPR2 - Mutation resultieren; in diesem Fall könnte Gremlin-1 helfen, die verringerte Aktivität des BMPRII-SMAD1/5/8-Wegs in Gegenwart von intaktem BMPR2 zu erklären
63
. Wichtig ist, dass die therapeutische Immunneutralisierung von Gremlin-1 den pulmonalen Gefäßumbau bei experimenteller PAH reduziert
65
. Also Umkehrung von Grem1Überexpression könnte ein Schlüsselmechanismus sein, durch den ActRIIA-Fc die SMAD1/5/8-Signalisierung mit der SMAD2/3-Signalisierung in den Lungengefäßen wieder ins Gleichgewicht bringt
28
. Da Gremlin-1 als Mediator der Hemmung des BMPRII-Signalwegs durch Endothelin
66
fungiert, könnte die Gremlin-1-Modulation auch ein potenzieller Punkt der mechanistischen Konvergenz zwischen den therapeutischen Wirkungen von ActRIIA-Fc und denen von Endothelin-Rezeptor-Antagonisten bei Patienten mit PAH sein.Der vaskuläre Umbau bei PAH wird allgemein so verstanden, dass er aus physiologischen zellulären Reaktionen auf Stress oder Verletzungen entsteht, die schließlich dysreguliert und anhaltend werden. Entzündung, Hypoxie oder biomechanischer Stress bei Personen mit eingeschränkter Aktivität des BMPRII-Signalwegs können eine pathologische Remodellierung der extrazellulären Matrix, abnormale Zellproliferation und möglicherweise EndMT fördern
67
. Jüngste Ergebnisse deuten darauf hin, dass BMPRII eine schützende Rolle bei der Homöostase von Endothelzellen spielt, wobei der Verlust von BMPRII EndMT begünstigt und die Zellen in einen vorbereiteten biomechanischen Zustand treibt, in dem Änderungen der Steifheit oder Scherspannung einen zweiten Schlag liefern und einen selbsterhaltenden Zyklus von überschüssigem TGF einleiten -β-Signalisierung
10
. Die chronische TGF-β1-Signalgebung
- Sachindra R. Joshi ,
- Jun Liu ,
- Troja Bloom ,
- Elif Karaca Atabay ,
- Tzu-Hsing Kuo ,
- Michael Lee ,
- Elitza Belcheva ,
- Matthäus Spaits ,
- Rosa Grenha ,
- Michelle C. Maguire ,
- Jeffrey L. Frost ,
- Kathryn Wang ,
- Steven D. Briscoe ,
- Mark J. Alexander ,
- Brantley R. Herrin ,
- Roselyne Castonguay ,
- R. Scott Pearsall ,
- Patrick André ,
- Paul B. Yu ,
- Ravindra Kumar &
- Gang Li
- 950 Zugriffe
- 3 Altmetrisch
- MetrikenEinzelheiten
. Die therapeutische Behandlung von SuHxNx-Ratten mit RAP-011 von Woche 5 bis Woche 9 übte eine robuste normalisierende Wirkung auf dieses pathologische Genexpressionsprofil aus (Abb.
1
C). Bis Woche 9 normalisierte die Behandlung mit RAP-011 die Expression von 207 von 248 (84 %) hochregulierten DEGs und 69 von 97 (71 %) herunterregulierten DEGs.
Im Gegensatz dazu veränderte die therapeutische Behandlung von SuHxNx
-Ratten mit Sildenafil die Expression von nur 27 von insgesamt 345 (8 %) DEGs ( 1C ). Die Hauptkomponentenanalyse ergab, dass Lungengewebe von mit RAP-011 behandelten SuHxNx-Ratten ein Genexpressionsprofil aufwies, das global normalem Gewebe ähnelte, während das Profil der mit Sildenafil behandelten SuHxNx-Rattenlungen in Woche 5 stärker dem der unbehandelten SuHxNx-Rattenlungen ähnelte (ergänzende Abb
2
_). Diese Ergebnisse weisen darauf hin, dass die therapeutische Behandlung mit RAP-011 eine deutliche korrigierende Wirkung auf das globale pathologische Genexpressionsprofil bei schwerer experimenteller PAH ausübt, die von der Behandlung mit einem Standard-Vasodilatator, der derzeit für die PAH-Therapie verfügbar ist, nicht erreicht wird.Anschließend verwendeten wir die Ingenuity Pathway Analysis (IPA), um dysregulierte Signalwege und potenzielle vorgeschaltete Regulatoren zu identifizieren, die mit allen DEGs (definiert durch den angepassten p-Wert < 0,001) assoziiert sind, basierend auf einem Vergleich des Lungengewebes in unbehandelten SuHxNx-Ratten sowohl in Woche 5 als auch in Woche 9 mit Lungengewebe bei normalen Ratten. Diese Analyse identifizierte 58 Signalwege, die in erkrankten Lungen sowohl in Woche 5 als auch in Woche 9 im Vergleich zu normalem Gewebe signifikant fehlreguliert waren. Gemäß der Rangfolge nach der Fisher-Methode umfassen die wichtigsten kanonischen Wege diejenigen, die Endothel- und Gefäßverletzungsreaktionen vermitteln (Gerinnungs-, Prothrombinaktivierungs- und Glykoprotein-VI-Wege); Entzündung und Immunantwort (Komplement, dendritische Zellreifung, Mustererkennung, Interleukin-10 und angeborene und adaptive Immunantwortwege); und TGF-β-Signalisierung (Tabelle
1
), die alle mit dem Fortschreiten der PAH bei Patienten oder präklinischen Modellen oder beidem in Verbindung gebracht wurden
2
,
4
. Interessanterweise war der wichtigste vorgeschaltete Regulator, der durch diese Analyse identifiziert wurde, der Tumornekrosefaktor (TNF), ein Schlüsselregulator von Entzündungs- und Immunantworten, der auch die Expression von BMPRII hemmt
37
. Andere hochrangige Upstream-Regulatoren wie VCAN, EPHB1, EGLN1 und TSC2 sind dafür bekannt, dass sie die Zellproliferation und -migration regulieren. Weitere Kandidaten für vorgeschaltete Regulatoren, die von IPA identifiziert wurden, umfassen TGFBR2 und Gene, die am Mitogen-aktivierten Proteinkinase-Weg beteiligt sind (ERK, p38MAPK und MAP2k1), gut charakterisierte Regulatoren von Entzündung und Zellproliferation (Tabelle
1
). Insgesamt zeigen die Signalwege und vorgeschalteten Regulatoren, die durch diese Analyse in der SuHxNx-Lungenpathologie involviert sind, einen Phänotyp einer hochgradig aktivierten Entzündung und Proliferation.Tabelle 1 Die am besten eingestuften Signalwege und vorgeschalteten Regulatoren gemäß Ingenuity Pathway Analysis in Lungen eines SuHxNx-Rattenmodells schwerer PAH.
Übereinstimmung bei abweichenden Genexpressionsprofilen zwischen einem Rattenmodell mit schwerer angioobliterativer PAH und PAH-PatientenAls nächstes untersuchten wir die potenzielle Relevanz dieser fehlregulierten Signalwege für die PAH-Pathogenese bei Patienten. Öffentlich zugängliche menschliche Transkriptomdaten, die von 58 PAH-Lungen und 25 Kontrolllungen (
www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117261
) gesammelt wurden, wurden IPA unterzogen, um signifikant fehlregulierte zu identifizieren Signalwege, die dann mit den in SuHxNx-Rattenlunge identifizierten Signalwegen verglichen wurden (Tabelle
1
). In Übereinstimmung mit veröffentlichten Humandaten umfassten die hier im Lungengewebe von PAH-Patienten identifizierten Signalwege an erster Stelle solche, die G-Protein-gekoppelte Rezeptoren beinhalten, und mehrere Signalwege, die mit Entzündungen und Immunantworten verbunden sind. Wichtig ist, dass IPA 18 Signalwege im Zusammenhang mit Entzündungs- und Immunantworten identifizierte, die im Lungengewebe von PAH-Patienten und dem SuHxNx-Rattenmodell für schwere PAH signifikant fehlreguliert sind (Tabelle
2
). Unter anderen solchen Signalwegen sind jene, die TGF-β- und BMP-Signalgebung vermitteln. Diese vom IPA erhaltenen Ergebnisse weisen auf eine Übereinstimmung in der fehlregulierten Genexpression zwischen dem SuHxNx-Rattenmodell und dem Lungengewebe von PAH-Patienten hin und liefern einen bestätigenden Beweis dafür, dass die hier verwendete SuHxNx-Ratte ein robustes Modell für menschliche PAH ist, insbesondere in Bezug auf ihre überschwängliche entzündliche Genexpressionssignatur.Tabelle 2 Häufigste Wege in der Lunge von SuHxNx-Ratten und PAH-Patienten.
ActRIIA-Fc zielt in verschiedenen PH-Modellen auf pulmonale Entzündungsmarker und Makrophageninfiltration abUm die Auswirkungen einer therapeutischen Behandlung mit entweder RAP-011 oder Sildenafil auf fehlregulierte Signalwege in der SuHxNx-Rattenlunge zu beurteilen, haben wir die relativen Expressionsniveaus von DEGs bestimmt, die mit diesen Signalwegen assoziiert sind. Die therapeutische Behandlung mit RAP-011 kehrte die anomale Expression vieler Gene in der Lunge um, die ansonsten im Zustand der SuHxNx-Krankheit aktiviert wurden (Abb. 1D
)
. Gene, die als Ergebnis der RAP-011-Behandlung differentiell exprimiert wurden, schlossen prominent jene in Signalwegen ein, die mit entzündlichen und abweichenden Immunantworten assoziiert sind ( 1D
)
. Andere DEGs sind mit dem Signalweg der TGF-β-Superfamilie assoziiert, insbesondere Grem1 (Gremlin-1), ein endogener BMP-Antagonist, der an der Proliferation von Endothelzellen und PAH beteiligt ist
38
. Mit der RAP-011-Behandlung verbundene Expressionsänderungen waren in den Fällen von Col2A1 , C6 und Grem1 besonders ausgeprägt . Im Gegensatz zu den auffälligen korrigierenden Wirkungen der RAP-011-Behandlung führte die therapeutische Behandlung mit Sildenafil zu begrenzten Veränderungen in der Expression von krankheitsassoziierten Genen (Abb.
1D
). Diese Ergebnisse liefern eine pulmonale Genexpressionssignatur, die den potenten therapeutischen Anti-Remodeling-Effekten von RAP-011 in diesem Modell schwerer PAH entspricht.Anschließend untersuchten wir die Auswirkungen der RAP-011-Therapie auf die Expression ausgewählter molekularer Entzündungs- und Immunmarker in der SuHxNx-Rattenlunge. Die PAH-Progression bei SuHxNx-Ratten war in Woche 9 mit einer signifikant erhöhten Expression von acht Schlüsselmarkern, einschließlich Il6 und Ccl2 , verbunden, und in jedem Fall normalisierte die therapeutische Behandlung mit RAP-011 – aber nicht Vehikel oder Sildenafil – ihre mRNA-Spiegel vollständig (Abb.
2
EIN). Wichtig ist, dass RAP-011 und Sildenafil in Kombination die Expression dieser Marker genauso effektiv normalisierten wie die RAP-011-Monotherapie (Abb. 2A
)
, was darauf hinweist, dass RAP-011 in diesem Modell einen robusten Nutzen bietet, selbst wenn es in Kombination mit einem Standard-Vasodilatator verwendet wird.Figur 2
wurden erhöhte hämodynamische Parameter wie der systolische Pulmonalarteriendruck (sPAP) und der mittlere Pulmonalarteriendruck (mPAP) effektiver durch eine duale Kombinationsbehandlung mit Antikörpern gegen Activin A/B und normalisiert GDF8/GDF11 als durch separate Antikörperbehandlungen (Abb.
3
C, D). Die duale Antikörperbehandlung normalisierte auch die RV-Hypertrophie effektiver als separate Antikörperbehandlungen (Abb.
3
E). Obwohl die Sequestrierung von Aktivinen oder GDFs in diesen Experimenten einen teilweisen Schutz vermittelte, implizieren unsere Ergebnisse, dass die Sequestrierung mehrerer Liganden des SMAD2/3-Wegs – wahrscheinlich Aktivine, GDF8 und GDF11 in Kombination – ein größeres Spektrum an therapeutischem Nutzen bietet und RAP-011 zugrunde liegt –induzierte Umkehrung kardiopulmonaler Beeinträchtigungen bei experimenteller PH.ActRIIA-Fc kehrt den Herzumbau bei schwerer experimenteller PAH umWir untersuchten die kardioprotektiven Wirkungen der RAP-011-Monotherapie weiter und verglichen sie mit Sildenafil im SuHxNx-Modell, das zu Beginn der therapeutischen Behandlung eine RV-Hypertrophie und einen beeinträchtigten Herzindex aufweist
28
(ergänzende Abb.
5
). Die therapeutische Behandlung mit RAP-011, beginnend 5 Wochen nach Krankheitsbeginn, verbesserte diese Parameter signifikant bis Woche 9 und führte zu einer größeren Verbesserung als die Behandlung mit Sildenafil (ergänzende Abb.
5
). Eine serielle Echokardiographie zeigte, dass die RAP-011-Therapie die RV-Dilatation, die Abflachung der Septumwand und die RV-Fraktionsflächenänderung (RVFAC) umkehrte, während Sildenafil dies nicht tat (Abb.
4
B, C). Wir bestätigten, dass die RAP-011-Therapie Anomalien der Pulmonalarterien-Beschleunigungszeit (PAAT) und der freien Wanddicke des RV (RVFWT) wirksamer linderte als Sildenafil (ergänzende Abb.
5
).Figur 4
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Bitte Anmelden oder Registrieren um der Konversation beizutreten.