
- Beiträge: 1755
Sidebar
PH, Fallbeispiele, Hintergründe
13 Mai 2022 22:44 - 13 Mai 2022 23:02 #1462
von danny
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
PH, Fallbeispiele, Hintergründe wurde erstellt von danny
www.rcpe.ac.uk/college/journal/pulmonary...-specialistPulmonale Hypertonie für den Laien Autor(en): Harrison Stubbs 1 , Martin Johnson 2Autorenverbindungen: 1 Pulmonary Vascular Fellow, 2 Respiratory and Pulmonary Vascular Consultant, Scottish Pulmonary Vascular Unit, Golden Jubilee National Hospital, Glasgow, UKKorrespondenz: Harrison Stubbs, Scottish Pulmonary Vascular Unit, Golden Jubilee National Hospital, Agamemnon St, Glasgow G81 4DY, UKE- Mail: Diese E-Mail-Adresse ist vor Spambots geschützt! Zur Anzeige muss JavaScript eingeschaltet sein!Zeitschriftenausgabe: Band 51: Ausgabe 4: 2021Papier zitieren als: JR Coll Physicians Edinb 2021; 51: 392–401
Format
Pulmonale Hypertonie ist eine seltene und komplexe Erkrankung, die aus einer Vielzahl von Grunderkrankungen entsteht. Die therapeutischen Möglichkeiten haben sich in den letzten zwei Jahrzehnten enorm erweitert, was zu erheblichen Verbesserungen der Prognose für einige Patienten geführt hat. Daher ist es wichtig, dass die Krankheit frühzeitig erkannt und zur weiteren Untersuchung und endgültigen Diagnose an spezialisierte Zentren überwiesen wird, um die Prognose bei diesem lebensverändernden Zustand zu verbessern. Leider ist es in Großbritannien immer noch so, dass die Diagnose der pulmonalen Hypertonie verzögert wird, oft Monate oder Jahre nach dem Auftreten der Symptome. Diese Übersicht zielt darauf ab, die wichtigsten Punkte bei der anfänglichen Behandlung und Überweisung von Patienten mit Verdacht auf pulmonale Hypertonie hervorzuheben, und stellt drei Fälle vor, um diese Bereiche zu unterstreichen.EinführungEine pulmonale Hypertonie (PH) liegt vor, wenn der mittlere pulmonalarterielle Druck (mPAP) ≥ 25 mmHg beträgt, gemessen während der Rechtsherzkatheterisierung (RHC). 1 Dies ist ein relativ willkürlicher Grenzwert, der ursprünglich gewählt wurde, um eine Überdiagnose und Behandlung zu verhindern. Im Jahr 2018 wurde ein überarbeiteter Schwellenwert von 20 mmHg vorgeschlagen, um PH zu definieren. Dies berücksichtigt eine systematische Überprüfung der mPAP-Bereiche für gesunde Personen, wobei ≥ 20 mmHg mehr als zwei Standardabweichungen über dem Bevölkerungsmittel entspricht. 2Klinische EinordnungPH wird in fünf klinische Gruppen mit ähnlicher Pathophysiologie, Hämodynamik und Ansprechen auf die Behandlung innerhalb jeder Gruppe eingeteilt. Diese sind in Tabelle 1 zusammengefasst.Tabelle 1 Klinische und hämodynamische Klassifikation der pulmonalen Hypertonie 2GruppeUntergruppePVR (WU)PAWP (mmHg)Gruppe IPulmonale arterielle Hypertonie (PAH)Idiopathisch (IPAH)Erblich (HPAH)Drogen- und Toxin-induziertBindegewebserkrankung (CTD-PAH)Portale Hypertonie (Portopulmonal, PoPAH)Angeborene Herzfehler (CHD-PAH)Lungenvenenverschlusskrankheit (PVOD)Andere: HIV, Bilharziose, persistierende PH des Neugeborenen≥3≤15Gruppe IILinksherzkrankheit (PH-LHD)Herzinsuffizienz mit erhaltener linksventrikulärer Ejektionsfraktion (HFpEF)Herzinsuffizienz mit reduzierter linksventrikulärer Ejektionsfraktion (HFrEF)HerzklappenerkrankungenIsolierte Postkapillare≤3>15Kombiniert Prä- und Postkapillare≥3>15 Gruppe IIILungenerkrankung und/oder HypoxieRestriktive LungenerkrankungObstruktive LungenerkrankungHypoxie ohne Lungenerkrankung ≥3≤15Gruppe IVPulmonale arterielle ObstruktionChronisch thromboembolische pulmonale Hypertonie (CTEPH)Andere Obstruktionen der Lungenarterie ≥3≤15Gruppe VMultifaktorielle oder unklare UrsachenHämatologische ErkrankungenSystemische und metabolische StörungenKomplexer angeborener Herzfehler ≥3≤15 Wie in Fall 1 gezeigt, ist die pulmonale arterielle Hypertonie (PAH, Gruppe I) eine primäre pulmonale Vaskulopathie, bei der ein Anstieg des pulmonalen Gefäßwiderstands (PVR) zu einer fortschreitenden rechtsventrikulären Belastung und einem Versagen führt. 3 PH aufgrund einer Linksherzerkrankung (LHD, Gruppe II) wird bei RHC durch einen pulmonalarteriellen Keildruck (PAWP) > 15 mmHg definiert und entsteht durch einen Anstieg des linksatrialen Drucks aufgrund einer zugrunde liegenden LHD. 4 PH aufgrund von Lungenerkrankungen und/oder Hypoxie (Gruppe III) tritt aufgrund von Mechanismen wie hypoxischer Vasokonstriktion und Obliteration des Lungengefäßbetts auf. 5PH als Folge einer Pulmonalarterienobstruktion (Gruppe IV) besteht hauptsächlich aus chronischer thromboembolischer pulmonaler Hypertonie (CTEPH). Dies wird durch die Nichtauflösung und Organisation von thromboembolischem Material nach einer Lungenembolie (LE) verursacht und ist bemerkenswert für die potenziell kurativen chirurgischen Behandlungsoptionen, die zur Verfügung stehen. 6 Gruppe V umfasst eine Mischung von Krankheiten mit multifaktoriellen oder unklaren Mechanismen. 2PH kann auch durch den Ort des Krankheitsprozesses in Bezug auf das pulmonale Gefäßkapillarbett definiert werden, wo die Gruppen I, III, IV und V typischerweise präkapillär auftreten. 2 Die Gruppen II und V können als isolierte postkapilläre (IpcPH) ohne intrinsische Erhöhung des PVR oder als kombinierte post- und präkapilläre (CpcPH) mit erhöhtem PVR vorliegen. 4Epidemiologie96 % der PH in Europa sind auf Linksherz- oder chronische Lungenerkrankungen zurückzuführen. 7 Im Vereinigten Königreich sind etwa 48–55 Personen pro Million (ppm) von PAH betroffen, mit einer Inzidenz von 6 ppm pro Jahr. Es wird anerkannt, dass die Inzidenz von CTEPH mit 3–6 ppm pro Jahr bei einer Prävalenz von 26–38 ppm zu wenig gemeldet wird. 8–10 Weltweit können die Ursachen von PH sehr unterschiedlich sein, wobei Fälle von PH im Zusammenhang mit Sichelzellenanämie und Infektionskrankheiten (HIV, Schistosomiasis, rheumatische Herzkrankheit nach Streptococcus ) häufiger in Ländern vorkommen, in denen diese noch endemisch sind. 7Im Vereinigten Königreich beträgt das Durchschnittsalter bei der Diagnose einer idiopathischen pulmonalen arteriellen Hypertonie (IPAH) 60 Jahre, und 37,7 % der IPAH-Patienten sind männlich. 11 In den letzten 20 Jahren sind neu diagnostizierte Patienten mit PAH mit größerer Wahrscheinlichkeit älter und haben mehrere Komorbiditäten, was die zunehmende Anerkennung von PH bei älteren Patienten und ihre Weiterverweisung widerspiegelt. 12,13PrognoseWährend PH eine fortschreitende und lebensbegrenzende Erkrankung bleibt, variiert die Prognose je nach Ätiologie und verfügbaren Behandlungen erheblich. 11 Im Vereinigten Königreich beträgt die 5-Jahres-Überlebensrate für Gruppe II 45 % und für Gruppe III 23 %. Allerdings haben sich die 5-Jahres-Überlebensraten für jüngere Patienten (18–53 Jahre) mit PAH im letzten Jahrzehnt von 74 % auf 83–85 % verbessert, und Patienten mit CTEPH, die sich einer chirurgischen Behandlung unterziehen, haben eine 5-Jahres-Überlebensrate von 85 %. 11,12 Bei PAH-Patienten, die auf eine Vasoreaktivitätsprovokation ansprechen und mit Calciumkanalblockern behandelt werden, schränkt PH ihre Lebenserwartung möglicherweise nicht ein.PathophysiologieBei Patienten mit PAH führen Umbau und Obstruktion der Lungengefäße zu einem erhöhten PVR. Wenn die Nachlast des rechten Ventrikels (RV) zunimmt, entwickelt sich eine Belastung des rechten Herzens mit Drucküberlastung, was dazu führt, dass der RV von einem Niederdruck- zu einem Hochdrucksystem umgestaltet wird. Als Reaktion auf die nachfolgende Volumenüberlastung erweitert sich der RV, um das Schlagvolumen aufrechtzuerhalten, und es entwickelt sich eine funktionelle Trikuspidalinsuffizienz (TR). Schließlich wird der RV dekompensieren, was zur Entwicklung einer Rechtsherzinsuffizienz führt. 14,15Abbildung 1 (a, c) zeigt Merkmale der RV-Dekompensation in einem Magnetresonanzbild des Herzens.Abbildung 1 Kardiales Magnetresonanzbild eines Patienten mit IPAH (siehe Fall 1), vor (a und c) und nach (b und d) der Behandlung mit einer krankheitsgerichteten Therapie. Die Tafeln (a) und (b) zeigen die Vierkammer-Querebene; (c) und (d) zeigen die Zweikammer-Sagittalebene. Die Felder (a) und (c) zeigen das Herz in der Enddiastole mit einem vergrößerten RV, der eine Kompression des LV mit Septumdeviation verursacht (Pfeil 1). Pfeil 2 bezeichnet einen Perikarderguss. Die Felder (b) und (d) zeigen enddiastolische Ansichten desselben Patienten nach der Behandlung, was auf eine Gesamtreduktion der Herzgröße, eine Verbesserung des Perikardergusses und eine Normalisierung der RV-Größe hinweist.
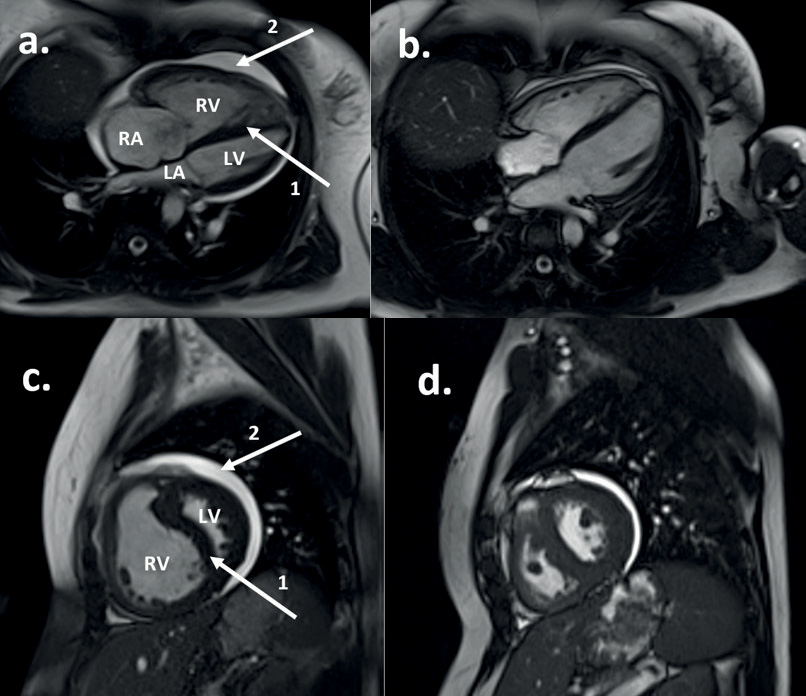
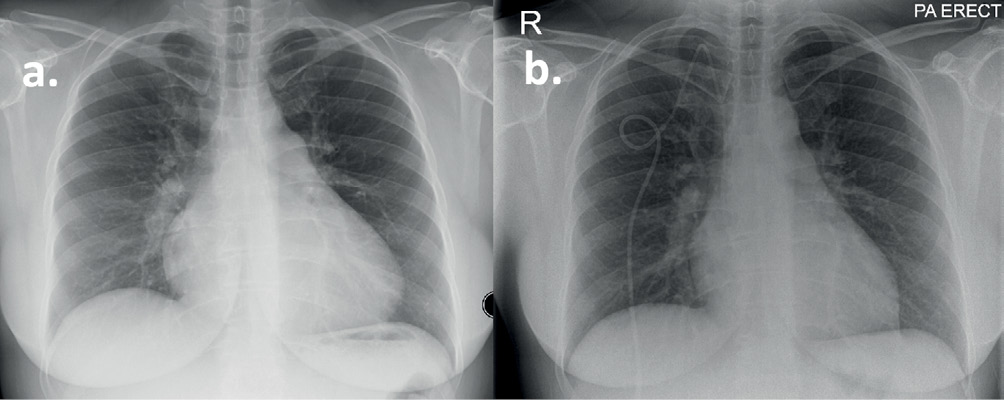
 Fall 1Eine 35-jährige Frau stellte sich nach einer Präsynkopen-Episode nach der Anwendung von Cyclizin gegen Übelkeit vor. Bei weiteren Befragungen litt sie seit einem Jahr an Atemnot bei Anstrengung und hatte kürzlich zwei synkopische Episoden erlebt. Transthorakales Echokardiogramm (TTE), das einen erhöhten systolischen Pulmonalarteriendruck (sPAP) von 134 mmHg und einen erweiterten und beeinträchtigten RV zeigte. Computertomographie-Lungenangiographie (CTPA) berichtete, dass sie eine bilaterale Lungenembolie mit Rechtsherzdilatation zeigt. Sie wurde mit Warfarin behandelt und anschließend entlassen. Trotz Antikoagulation schritten ihre Symptome fort und sie entwickelte Brustschmerzen bei Belastung und eine Verschlechterung der Präsynkope, was zu einer erneuten Aufnahme und Verlegung in die Scottish Pulmonary Vascular Unit (SPVU) mit einer vorläufigen Diagnose von CTEPH führte. Spirometrie wurde beibehalten, DLCO (Transferfaktor für Kohlenmonoxid) war um 53 % des Sollwerts reduziert, NTproBNP war nicht erhöht. Sie ging 426 m bei einem Sechs-Minuten-Gehtest (6 MWT) und war leicht entsättigt zu einem SpO2von 94 %. RHC zeigte einen erhöhten mPAP von 71 mmHg, einen normalen PAWP von 9 mmHg und einen erhöhten PVR von 17,2 Wood-Einheiten (WU). Die radiologische Überprüfung des anfänglichen CTPA stimmte nicht mit der Diagnose einer akuten LE überein, und das Lungenangiogramm, das Magnetresonanz-Lungenangiogramm (MRPA) und das wiederholte CTPA zeigten auch keine Merkmale einer chronischen oder akuten pulmonalen thromboembolischen Erkrankung. Aufgrund des Fehlens einer zugrunde liegenden Ätiologie wurde ihre Diagnose auf IPAH geändert. Aufgrund der Vorgeschichte synkopaler Episoden wurde bei ihr eine krankheitsgerichtete Dreifachtherapie mit i.v. Epoprostenol, Sildenafil und Macitentan begonnen. Sie hatte danach eine deutliche Verbesserung ihrer Symptome, ist wieder arbeitsfähig und gehört jetzt der Funktionsklasse I der Weltgesundheitsorganisation (WHO FC) an. Nach sechsjähriger Behandlung mit i.v. Epoprostenol entwickelte sie eine Hickman-Linien-Infektion Aufforderung zur Überprüfung ihrer Therapien. Das i.v. Epoprostenol wurde durch orales Selexipag ohne Anzeichen einer Verschlechterung ersetzt (Abbildungen 1 und 2).Abbildung 2 Röntgen-Thorax eines Patienten mit IPAH (siehe Fall 1). (a) Postero-anteriore Projektion bei der Diagnose, die eine Kardiomegalie und erweiterte Pulmonalarterien zeigt. (b) Nach sechsjähriger Behandlung mit einer krankheitsgerichteten Dreifachtherapie, die einen rechtsseitigen zentralvenösen Tunnelkatheter zur Verabreichung von Epoprostenol und eine Verbesserung der Herzgröße zeigt.
Fall 1Eine 35-jährige Frau stellte sich nach einer Präsynkopen-Episode nach der Anwendung von Cyclizin gegen Übelkeit vor. Bei weiteren Befragungen litt sie seit einem Jahr an Atemnot bei Anstrengung und hatte kürzlich zwei synkopische Episoden erlebt. Transthorakales Echokardiogramm (TTE), das einen erhöhten systolischen Pulmonalarteriendruck (sPAP) von 134 mmHg und einen erweiterten und beeinträchtigten RV zeigte. Computertomographie-Lungenangiographie (CTPA) berichtete, dass sie eine bilaterale Lungenembolie mit Rechtsherzdilatation zeigt. Sie wurde mit Warfarin behandelt und anschließend entlassen. Trotz Antikoagulation schritten ihre Symptome fort und sie entwickelte Brustschmerzen bei Belastung und eine Verschlechterung der Präsynkope, was zu einer erneuten Aufnahme und Verlegung in die Scottish Pulmonary Vascular Unit (SPVU) mit einer vorläufigen Diagnose von CTEPH führte. Spirometrie wurde beibehalten, DLCO (Transferfaktor für Kohlenmonoxid) war um 53 % des Sollwerts reduziert, NTproBNP war nicht erhöht. Sie ging 426 m bei einem Sechs-Minuten-Gehtest (6 MWT) und war leicht entsättigt zu einem SpO2von 94 %. RHC zeigte einen erhöhten mPAP von 71 mmHg, einen normalen PAWP von 9 mmHg und einen erhöhten PVR von 17,2 Wood-Einheiten (WU). Die radiologische Überprüfung des anfänglichen CTPA stimmte nicht mit der Diagnose einer akuten LE überein, und das Lungenangiogramm, das Magnetresonanz-Lungenangiogramm (MRPA) und das wiederholte CTPA zeigten auch keine Merkmale einer chronischen oder akuten pulmonalen thromboembolischen Erkrankung. Aufgrund des Fehlens einer zugrunde liegenden Ätiologie wurde ihre Diagnose auf IPAH geändert. Aufgrund der Vorgeschichte synkopaler Episoden wurde bei ihr eine krankheitsgerichtete Dreifachtherapie mit i.v. Epoprostenol, Sildenafil und Macitentan begonnen. Sie hatte danach eine deutliche Verbesserung ihrer Symptome, ist wieder arbeitsfähig und gehört jetzt der Funktionsklasse I der Weltgesundheitsorganisation (WHO FC) an. Nach sechsjähriger Behandlung mit i.v. Epoprostenol entwickelte sie eine Hickman-Linien-Infektion Aufforderung zur Überprüfung ihrer Therapien. Das i.v. Epoprostenol wurde durch orales Selexipag ohne Anzeichen einer Verschlechterung ersetzt (Abbildungen 1 und 2).Abbildung 2 Röntgen-Thorax eines Patienten mit IPAH (siehe Fall 1). (a) Postero-anteriore Projektion bei der Diagnose, die eine Kardiomegalie und erweiterte Pulmonalarterien zeigt. (b) Nach sechsjähriger Behandlung mit einer krankheitsgerichteten Dreifachtherapie, die einen rechtsseitigen zentralvenösen Tunnelkatheter zur Verabreichung von Epoprostenol und eine Verbesserung der Herzgröße zeigt.
 Klinische UntersuchungNach wie vor werden Patienten mit PH erst spät im Krankheitsverlauf diagnostiziert (Kasten 1). 16–18 Um die Diagnose zu erleichtern, sind grundlegende Kenntnisse des Zustands und ein Verdachtsindex erforderlich. Tabelle 2 zeigt, wie sich Patienten mit PH bei einer Vielzahl von Fachgebieten der Sekundärversorgung vorstellen können.Box 1 Warum gibt es eine Verzögerung bei der Diagnose von PH?
Klinische UntersuchungNach wie vor werden Patienten mit PH erst spät im Krankheitsverlauf diagnostiziert (Kasten 1). 16–18 Um die Diagnose zu erleichtern, sind grundlegende Kenntnisse des Zustands und ein Verdachtsindex erforderlich. Tabelle 2 zeigt, wie sich Patienten mit PH bei einer Vielzahl von Fachgebieten der Sekundärversorgung vorstellen können.Box 1 Warum gibt es eine Verzögerung bei der Diagnose von PH?
 Kasten 2 Andere echokardiographische Zeichen einer pulmonalen HypertonieEin TRPG im Bereich von 32–45 mmHg mit Anzeichen aus zwei der folgenden Kategorien (A/B/C) sollte zu weiteren Untersuchungen und Überweisungen führen.
Kasten 2 Andere echokardiographische Zeichen einer pulmonalen HypertonieEin TRPG im Bereich von 32–45 mmHg mit Anzeichen aus zwei der folgenden Kategorien (A/B/C) sollte zu weiteren Untersuchungen und Überweisungen führen.
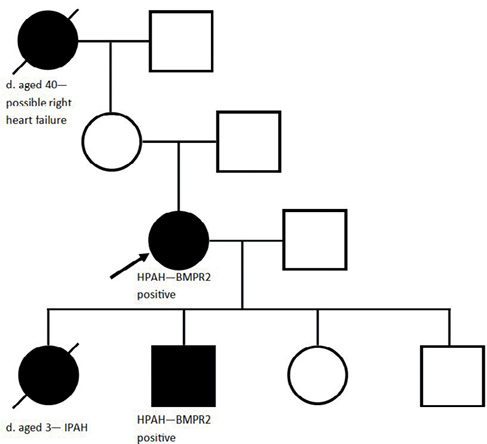
 Da Patienten mit HPAH, am häufigsten mit einer monoallelischen Mutation im BMPR2 -Gen, in einem jüngeren Alter mit schlechterer Prognose vorstellig werden, sollte bei allen Patienten mit IPAH ein genetisches Screening in Betracht gezogen werden, auch wenn keine Familienanamnese vorliegt. Fünfundzwanzig Prozent der IPAH-Patienten haben eine BMPR2 - Mutation und dies eröffnet die Möglichkeit eines genetischen Screenings für Familienmitglieder, falls positiv, 26,27 , wie in Fall 2 gezeigt. Patienten mit Merkmalen von PVOD können für Mutationen im EIF2AK4 -Gen homozygot sein , was eine diagnostische Bestätigung dieses Zustands ermöglichen kann. 26Fall 2Eine 41-jährige Frau und derzeitige Raucherin wurde an die SPVU überwiesen, die sich seit zwei Jahren in Übereinstimmung mit WHO FC III mit allmählich zunehmender Atemnot bei Anstrengung verschlechterte. Ihre Großmutter war im Alter von 40 Jahren an „Wassersucht“ gestorben, und ihre Tochter war im Alter von drei Jahren nach einer kurzlebigen Krankheit gestorben, die anschließend bei der Obduktion als IPAH diagnostiziert wurde. Bei der Untersuchung hatte der Patient einen lauten P2, ein RV-Hebeln, war zentral cyanosiert und in Ruhe systemisch hypotensiv (93/67 mmHg). NTproBNP war nicht erhöht und sie ging 331 m auf einem 6MGT, leicht entsättigt auf einen SpO 2von 94 %. TTE zeigte einen TRPG von 55 mmHg und einen stark erweiterten RV. Die CT-Bildgebung zeigte eine Dilatation der Hauptpulmonalarterie und des RV, ein leichtes Emphysem und keine Anzeichen einer thrombotischen Erkrankung. RHC zeigte einen erhöhten mPAP von 59 mmHg, einen normalen PAWP von 8 mmHg und einen sehr hohen PVR von 22,2 WU. Mit der positiven Familienanamnese stimmten diese Ergebnisse mit einer HPAH-Diagnose überein. Es gab Bedenken hinsichtlich der Behandlung mit einem Phosphodiesterase-5-Hemmer aufgrund einer Retinitis pigmentosa in der Familienanamnese, und daher wurde eine Behandlung mit Bosentan begonnen. Riociguat wurde sechs Jahre nach der Diagnose hinzugefügt. Sie bleibt in diesen zehn Jahren nach der Diagnose stabil und ist jetzt WHO FC II. Angesichts der Familienanamnese wurden ihre verbleibenden drei Kinder mit TTE gescreent. Ihr 14-jähriger Sohn hatte einen erhöhten TRPG von 61 mmHg und ließ sich im RHC HPAH bestätigen. Er bleibt gut in FC II unter Bosentan-Monotherapie. Eine anschließende genetische Analyse ergab, dass die Patientin und ihr Sohn positiv auf eine monoallelische Mutation in der Mutation warenBMPR2 -Gen (Abbildung 5).Abbildung 5 Genetischer Stammbaum einer Patientin mit HPAH (siehe Fall 2). Der schwarze Pfeil zeigt den Indexpatienten an. Ein gefülltes schwarzes Kästchen zeigt eine Person an, die von PH betroffen ist, und eine diagonale Linie zeigt an, dass eine Person verstorben ist (d. zeigt das Alter beim Tod an).
Da Patienten mit HPAH, am häufigsten mit einer monoallelischen Mutation im BMPR2 -Gen, in einem jüngeren Alter mit schlechterer Prognose vorstellig werden, sollte bei allen Patienten mit IPAH ein genetisches Screening in Betracht gezogen werden, auch wenn keine Familienanamnese vorliegt. Fünfundzwanzig Prozent der IPAH-Patienten haben eine BMPR2 - Mutation und dies eröffnet die Möglichkeit eines genetischen Screenings für Familienmitglieder, falls positiv, 26,27 , wie in Fall 2 gezeigt. Patienten mit Merkmalen von PVOD können für Mutationen im EIF2AK4 -Gen homozygot sein , was eine diagnostische Bestätigung dieses Zustands ermöglichen kann. 26Fall 2Eine 41-jährige Frau und derzeitige Raucherin wurde an die SPVU überwiesen, die sich seit zwei Jahren in Übereinstimmung mit WHO FC III mit allmählich zunehmender Atemnot bei Anstrengung verschlechterte. Ihre Großmutter war im Alter von 40 Jahren an „Wassersucht“ gestorben, und ihre Tochter war im Alter von drei Jahren nach einer kurzlebigen Krankheit gestorben, die anschließend bei der Obduktion als IPAH diagnostiziert wurde. Bei der Untersuchung hatte der Patient einen lauten P2, ein RV-Hebeln, war zentral cyanosiert und in Ruhe systemisch hypotensiv (93/67 mmHg). NTproBNP war nicht erhöht und sie ging 331 m auf einem 6MGT, leicht entsättigt auf einen SpO 2von 94 %. TTE zeigte einen TRPG von 55 mmHg und einen stark erweiterten RV. Die CT-Bildgebung zeigte eine Dilatation der Hauptpulmonalarterie und des RV, ein leichtes Emphysem und keine Anzeichen einer thrombotischen Erkrankung. RHC zeigte einen erhöhten mPAP von 59 mmHg, einen normalen PAWP von 8 mmHg und einen sehr hohen PVR von 22,2 WU. Mit der positiven Familienanamnese stimmten diese Ergebnisse mit einer HPAH-Diagnose überein. Es gab Bedenken hinsichtlich der Behandlung mit einem Phosphodiesterase-5-Hemmer aufgrund einer Retinitis pigmentosa in der Familienanamnese, und daher wurde eine Behandlung mit Bosentan begonnen. Riociguat wurde sechs Jahre nach der Diagnose hinzugefügt. Sie bleibt in diesen zehn Jahren nach der Diagnose stabil und ist jetzt WHO FC II. Angesichts der Familienanamnese wurden ihre verbleibenden drei Kinder mit TTE gescreent. Ihr 14-jähriger Sohn hatte einen erhöhten TRPG von 61 mmHg und ließ sich im RHC HPAH bestätigen. Er bleibt gut in FC II unter Bosentan-Monotherapie. Eine anschließende genetische Analyse ergab, dass die Patientin und ihr Sohn positiv auf eine monoallelische Mutation in der Mutation warenBMPR2 -Gen (Abbildung 5).Abbildung 5 Genetischer Stammbaum einer Patientin mit HPAH (siehe Fall 2). Der schwarze Pfeil zeigt den Indexpatienten an. Ein gefülltes schwarzes Kästchen zeigt eine Person an, die von PH betroffen ist, und eine diagonale Linie zeigt an, dass eine Person verstorben ist (d. zeigt das Alter beim Tod an).
 ManagementEs hat sich gezeigt, dass eine frühzeitige medikamentöse Kombinationstherapie die Ergebnisse bei PAH verbessert. 28 Gegenwärtige pharmakologische Therapien zielen auf drei unterschiedliche biologische Stoffwechselwege ab: den Stickoxid-, Endothelin- und Prostanoid-Weg, der in Tabelle 4 weiter ausgeführt wird. 29,30 Aktuelle Leitlinien befürworten eine anfängliche Kombinationstherapie, sofern nicht kontraindiziert. 30 Bei Patienten mit schwererer Erkrankung oder bei denen sich der Zustand nach der Erstbehandlung nicht bessert, kann eine parenterale Behandlung erforderlich sein, z. B. mit Epoprostenol über einen zentralvenösen Tunnelkatheter. 29 Eine Untergruppe von IPAH zeigt eine hämodynamische Verbesserung mit pulmonalen Vasoreaktivitätstests während RHC und kann mit Kalziumkanalblockern behandelt werden, oft mit dramatischer klinischer Verbesserung. 19Tabelle 4 Therapeutische Ziele bei pulmonaler HypertonieWegBeispiele für MedikamenteVerwaltungDosisNebenwirkungen und ÜberwachungStickoxid und cGMP-WegPhosphodiesterase-5-Inhibitoren (PDE5i)SildenafilOral, intravenösDreimal täglich – variable Dosierung*Verdauungsstörungen, Sodbrennen, Kopfschmerzen, Gesichtsrötung, Kieferschmerzen, Hypotonie, Sehbehinderung, Hörbehinderung, PriapismusTadalafilOralEinmal täglich – 20 mg, 40 mgLösliche Guanylatcyclase (sGC)-AktivatorenRiociguatOral1–2,5 mg dreimal täglichHypotonieKontraindiziert mit PDE5iEndothelin-Rezeptor-AntagonistenBosentanOral62,5–125 mg zweimal täglichLeberfunktionsteststörung und Anämie (Blutüberwachung erforderlich)TeratogenKopfschmerzen, Hitzewallungen, Hypotonie, FlüssigkeitsretentionMacitentan 10 mg einmal täglichAmbrisentan 5–10 mg einmal täglichProstanoide und cAMP-WegIloprostVernebeltIntravenös10–20 μg siebenmal täglichKontinuierliche InfusionSchwierigkeiten mit VerneblerFlush, Trismus, HypotonieSelexipagOral200–1.600 μg zweimal täglichHitzewallungen, Durchfall, Übelkeit, ErbrechenTreprostenilSubkutan, intravenös Komplikationen durch subkutanen ZugangEpoprostenolIntravenösKontinuierliche Infusion – beginnend mit 2 ng/kg/min und in Intervallen steigerndKomplikationen durch venösen ZugangHypotonie, Gesichtsrötung, Kieferschmerzen, Durchfall, Gliederschmerzen *Die zugelassene Dosis von Sildenafil beträgt 20 mg dreimal täglich, es können jedoch nicht zugelassene Dosen verwendet werden, einschließlich 25 mg, 50 mg, 75 mg und 100 mg dreimal täglich.Die Beurteilung der klinischen Verschlechterung und der Notwendigkeit einer Therapieeskalation basiert auf der Risikostratifizierung und umfasst die Diagnose des Patienten, die körperliche Leistungsfähigkeit, die FC der WHO, die hämodynamischen Werte, den NTproBNP/BNP-Assay und bildgebende Befunde, wie sie in Scores wie dem REVEAL 2.0-Risikorechner oder ausgedrückt werden Risikotabelle der European Respiratory Society/European Society of Cardiology. 30,31Die Behandlungsoptionen für alle Patienten sollten eine optimale Diurese, eine geeignete Impfung wie bei jeder chronischen Atemwegserkrankung und die Erwägung einer langfristigen Sauerstoffgabe bei Atemversagen umfassen. Betablocker und Cyclizin sollten vermieden werden, da sie eine Verringerung des Herzzeitvolumens verursachen. Nitrate sind zusammen mit Phosphodiesterase-5-Hemmern kontraindiziert und können bei Patienten, die diese einnehmen, nicht zur Behandlung von Angina pectoris verwendet werden. Wie in Fall 3 gezeigt, sollten weibliche PH-Patientinnen über die hohe Schwangerschaftssterblichkeit aufgeklärt und zu Verhütung und Schwangerschaftsabbruch beraten werden. 32Zustände, die eine PH verschlimmern, wie Rhythmusstörungen, Infektionen und akute Lungenembolie, sollten sofort erkannt und behandelt werden. Patienten mit akutem RV-Versagen müssen möglicherweise in einer Umgebung mit hoher Abhängigkeit mit inotroper und vasopressorischer Unterstützung behandelt werden. 33 Eine bilaterale Lungentransplantation wird bei Patienten in Erwägung gezogen, die auf eine optimale Therapie nicht ansprechen oder deren Zustand sich während der Therapie verschlechtert. 19 Aerobic-Übungen mit niedriger Intensität sind sicher und sollten bei Patienten mit PH ermutigt werden, um die Dekonditionierung zu reduzieren und die Lebensqualität der Patienten zu verbessern. 34 Patienten sollten nicht angewiesen werden, körperliche Betätigung zu vermeiden, sollten jedoch vor Aktivitäten gewarnt werden, die zu Symptomen einer Präsynkope oder Synkope führen.Wie in Fall 3 gezeigt, sollten Patienten mit CTEPH zur Erwägung einer chirurgischen Behandlung überwiesen werden. 6 In Großbritannien wird dies im Royal Papworth Hospital in Cambridge durchgeführt. Zu den chirurgischen Optionen gehören die pulmonale Endarteriektomie (PEA) bei einer proximalen Erkrankung und die Ballon-Lungenangioplastie bei einer distaleren Erkrankung. Während das Ansprechen auf PEA ausgezeichnet sein kann, geht es mit einem hypothermischen Kreislaufstillstand einher und birgt ein signifikantes perioperatives Mortalitätsrisiko von 2–5 %. 35 CTEPH-Patienten benötigen eine lebenslange Antikoagulation. 6Es gibt keine eindeutigen Hinweise auf eine Antikoagulation bei anderen Formen der präkapillären PH; Es ist jedoch zu beachten, dass bei CTD-PAH eine Antikoagulation aufgrund eines erhöhten Blutungsrisikos schädlich sein kann und nur bei eindeutiger Indikation begonnen werden sollte. 36Fall 3Eine zuvor gesunde 24-jährige Frau stellte sich akut mit einer einwöchigen Vorgeschichte von Atemnot bei Anstrengung nach einem Kurzstreckenflug vor drei Wochen vor. CTPA bestätigte ausgedehnte bilaterale Lungenembolien und sie wurde anschließend auf Rivaroxaban entlassen. Drei Monate später wurde sie schwanger und ihre Antikoagulation wurde auf niedermolekulares Heparin umgestellt. Während eines vorgeburtlichen Besuchs wurde festgestellt, dass sie bei Anstrengung atemlos war (WHO FC II), was eine Untersuchung mit TTE veranlasste, die eine reduzierte Pulmonalklappenbeschleunigungszeit von 73 ms (NR > 105 ms), eine leichte rechtsventrikuläre systolische Dysfunktion und eine RV-Dilatation zeigte. ohne sichtbare TR-Düse. Ein Beatmungs-Perfusions-Scan (V/Q) zeigte mehrere große, nicht übereinstimmende Perfusionsdefekte (Abbildung 6). Sie wurde an die SPVU überwiesen, zu diesem Zeitpunkt war ihr Fötus in der 12. Schwangerschaftswoche. Sie ging 470 m auf einem 6MGT und war deutlich entsättigt auf einen SpO2 von 80 %. Sie unterzog sich einer RHC und zeigte einen erhöhten mPAP von 40 mmHg, einen normalen PAWP von 6 mmHg und einen erhöhten PVR von 6,4 WU. Weder MRPA noch konventionelles Lungenangiogramm wurden aufgrund einer Schwangerschaft durchgeführt. Wiederholte CTPA zeigte Merkmale einer chronischen thromboembolischen Erkrankung (Mosaik, Bronchialarterienhypertrophie und Netz- und Verschlusskrankheit) im Einklang mit einer Diagnose von CTEPH (Abbildung 7). Sie wurde mit einer medizinischen Behandlung mit Sildenafil begonnen und an das Royal Papworth Hospital überwiesen, das darauf hinwies, dass PEA während der Schwangerschaft vermieden werden sollte. Die Patientin wurde über die Risiken einer Schwangerschaft bei PH beraten, aber gegen einen Schwangerschaftsabbruch gewählt. Unglücklicherweise trat in der 26. Schwangerschaftswoche der intrauterine Tod auf und die Patientin unterzog sich einer ereignislosen vaginalen Entbindung von fötalen Produkten. Tests zu diesem Zeitpunkt zeigten, dass sie dreifach positiv für das Antiphospholipid-Syndrom war, und ihre Antikoagulation wurde auf Warfarin umgestellt. Sie unterzog sich 16 Monate nach ihrer anfänglichen akuten Lungenembolie einer PEA am Royal Papworth Hospital, und die Nachsorge-RHC sechs Monate später bestätigte eine hämodynamische Verbesserung mit einem normalen mPAP von 23 mmHg, PAWP 11 mmHg und PVR 2,0 WU. Sildenafil wurde postoperativ abgesetzt. Die Patientin hatte anschließend eine erfolgreiche Schwangerschaft und setzt die lebenslange Antikoagulation fort.Abbildung 6 Beatmungs- und Perfusionsscan (V/Q) bei einem Patienten mit CTEPH (siehe Fall 3), der mehrere nicht übereinstimmende Perfusionsdefekte zeigt.
ManagementEs hat sich gezeigt, dass eine frühzeitige medikamentöse Kombinationstherapie die Ergebnisse bei PAH verbessert. 28 Gegenwärtige pharmakologische Therapien zielen auf drei unterschiedliche biologische Stoffwechselwege ab: den Stickoxid-, Endothelin- und Prostanoid-Weg, der in Tabelle 4 weiter ausgeführt wird. 29,30 Aktuelle Leitlinien befürworten eine anfängliche Kombinationstherapie, sofern nicht kontraindiziert. 30 Bei Patienten mit schwererer Erkrankung oder bei denen sich der Zustand nach der Erstbehandlung nicht bessert, kann eine parenterale Behandlung erforderlich sein, z. B. mit Epoprostenol über einen zentralvenösen Tunnelkatheter. 29 Eine Untergruppe von IPAH zeigt eine hämodynamische Verbesserung mit pulmonalen Vasoreaktivitätstests während RHC und kann mit Kalziumkanalblockern behandelt werden, oft mit dramatischer klinischer Verbesserung. 19Tabelle 4 Therapeutische Ziele bei pulmonaler HypertonieWegBeispiele für MedikamenteVerwaltungDosisNebenwirkungen und ÜberwachungStickoxid und cGMP-WegPhosphodiesterase-5-Inhibitoren (PDE5i)SildenafilOral, intravenösDreimal täglich – variable Dosierung*Verdauungsstörungen, Sodbrennen, Kopfschmerzen, Gesichtsrötung, Kieferschmerzen, Hypotonie, Sehbehinderung, Hörbehinderung, PriapismusTadalafilOralEinmal täglich – 20 mg, 40 mgLösliche Guanylatcyclase (sGC)-AktivatorenRiociguatOral1–2,5 mg dreimal täglichHypotonieKontraindiziert mit PDE5iEndothelin-Rezeptor-AntagonistenBosentanOral62,5–125 mg zweimal täglichLeberfunktionsteststörung und Anämie (Blutüberwachung erforderlich)TeratogenKopfschmerzen, Hitzewallungen, Hypotonie, FlüssigkeitsretentionMacitentan 10 mg einmal täglichAmbrisentan 5–10 mg einmal täglichProstanoide und cAMP-WegIloprostVernebeltIntravenös10–20 μg siebenmal täglichKontinuierliche InfusionSchwierigkeiten mit VerneblerFlush, Trismus, HypotonieSelexipagOral200–1.600 μg zweimal täglichHitzewallungen, Durchfall, Übelkeit, ErbrechenTreprostenilSubkutan, intravenös Komplikationen durch subkutanen ZugangEpoprostenolIntravenösKontinuierliche Infusion – beginnend mit 2 ng/kg/min und in Intervallen steigerndKomplikationen durch venösen ZugangHypotonie, Gesichtsrötung, Kieferschmerzen, Durchfall, Gliederschmerzen *Die zugelassene Dosis von Sildenafil beträgt 20 mg dreimal täglich, es können jedoch nicht zugelassene Dosen verwendet werden, einschließlich 25 mg, 50 mg, 75 mg und 100 mg dreimal täglich.Die Beurteilung der klinischen Verschlechterung und der Notwendigkeit einer Therapieeskalation basiert auf der Risikostratifizierung und umfasst die Diagnose des Patienten, die körperliche Leistungsfähigkeit, die FC der WHO, die hämodynamischen Werte, den NTproBNP/BNP-Assay und bildgebende Befunde, wie sie in Scores wie dem REVEAL 2.0-Risikorechner oder ausgedrückt werden Risikotabelle der European Respiratory Society/European Society of Cardiology. 30,31Die Behandlungsoptionen für alle Patienten sollten eine optimale Diurese, eine geeignete Impfung wie bei jeder chronischen Atemwegserkrankung und die Erwägung einer langfristigen Sauerstoffgabe bei Atemversagen umfassen. Betablocker und Cyclizin sollten vermieden werden, da sie eine Verringerung des Herzzeitvolumens verursachen. Nitrate sind zusammen mit Phosphodiesterase-5-Hemmern kontraindiziert und können bei Patienten, die diese einnehmen, nicht zur Behandlung von Angina pectoris verwendet werden. Wie in Fall 3 gezeigt, sollten weibliche PH-Patientinnen über die hohe Schwangerschaftssterblichkeit aufgeklärt und zu Verhütung und Schwangerschaftsabbruch beraten werden. 32Zustände, die eine PH verschlimmern, wie Rhythmusstörungen, Infektionen und akute Lungenembolie, sollten sofort erkannt und behandelt werden. Patienten mit akutem RV-Versagen müssen möglicherweise in einer Umgebung mit hoher Abhängigkeit mit inotroper und vasopressorischer Unterstützung behandelt werden. 33 Eine bilaterale Lungentransplantation wird bei Patienten in Erwägung gezogen, die auf eine optimale Therapie nicht ansprechen oder deren Zustand sich während der Therapie verschlechtert. 19 Aerobic-Übungen mit niedriger Intensität sind sicher und sollten bei Patienten mit PH ermutigt werden, um die Dekonditionierung zu reduzieren und die Lebensqualität der Patienten zu verbessern. 34 Patienten sollten nicht angewiesen werden, körperliche Betätigung zu vermeiden, sollten jedoch vor Aktivitäten gewarnt werden, die zu Symptomen einer Präsynkope oder Synkope führen.Wie in Fall 3 gezeigt, sollten Patienten mit CTEPH zur Erwägung einer chirurgischen Behandlung überwiesen werden. 6 In Großbritannien wird dies im Royal Papworth Hospital in Cambridge durchgeführt. Zu den chirurgischen Optionen gehören die pulmonale Endarteriektomie (PEA) bei einer proximalen Erkrankung und die Ballon-Lungenangioplastie bei einer distaleren Erkrankung. Während das Ansprechen auf PEA ausgezeichnet sein kann, geht es mit einem hypothermischen Kreislaufstillstand einher und birgt ein signifikantes perioperatives Mortalitätsrisiko von 2–5 %. 35 CTEPH-Patienten benötigen eine lebenslange Antikoagulation. 6Es gibt keine eindeutigen Hinweise auf eine Antikoagulation bei anderen Formen der präkapillären PH; Es ist jedoch zu beachten, dass bei CTD-PAH eine Antikoagulation aufgrund eines erhöhten Blutungsrisikos schädlich sein kann und nur bei eindeutiger Indikation begonnen werden sollte. 36Fall 3Eine zuvor gesunde 24-jährige Frau stellte sich akut mit einer einwöchigen Vorgeschichte von Atemnot bei Anstrengung nach einem Kurzstreckenflug vor drei Wochen vor. CTPA bestätigte ausgedehnte bilaterale Lungenembolien und sie wurde anschließend auf Rivaroxaban entlassen. Drei Monate später wurde sie schwanger und ihre Antikoagulation wurde auf niedermolekulares Heparin umgestellt. Während eines vorgeburtlichen Besuchs wurde festgestellt, dass sie bei Anstrengung atemlos war (WHO FC II), was eine Untersuchung mit TTE veranlasste, die eine reduzierte Pulmonalklappenbeschleunigungszeit von 73 ms (NR > 105 ms), eine leichte rechtsventrikuläre systolische Dysfunktion und eine RV-Dilatation zeigte. ohne sichtbare TR-Düse. Ein Beatmungs-Perfusions-Scan (V/Q) zeigte mehrere große, nicht übereinstimmende Perfusionsdefekte (Abbildung 6). Sie wurde an die SPVU überwiesen, zu diesem Zeitpunkt war ihr Fötus in der 12. Schwangerschaftswoche. Sie ging 470 m auf einem 6MGT und war deutlich entsättigt auf einen SpO2 von 80 %. Sie unterzog sich einer RHC und zeigte einen erhöhten mPAP von 40 mmHg, einen normalen PAWP von 6 mmHg und einen erhöhten PVR von 6,4 WU. Weder MRPA noch konventionelles Lungenangiogramm wurden aufgrund einer Schwangerschaft durchgeführt. Wiederholte CTPA zeigte Merkmale einer chronischen thromboembolischen Erkrankung (Mosaik, Bronchialarterienhypertrophie und Netz- und Verschlusskrankheit) im Einklang mit einer Diagnose von CTEPH (Abbildung 7). Sie wurde mit einer medizinischen Behandlung mit Sildenafil begonnen und an das Royal Papworth Hospital überwiesen, das darauf hinwies, dass PEA während der Schwangerschaft vermieden werden sollte. Die Patientin wurde über die Risiken einer Schwangerschaft bei PH beraten, aber gegen einen Schwangerschaftsabbruch gewählt. Unglücklicherweise trat in der 26. Schwangerschaftswoche der intrauterine Tod auf und die Patientin unterzog sich einer ereignislosen vaginalen Entbindung von fötalen Produkten. Tests zu diesem Zeitpunkt zeigten, dass sie dreifach positiv für das Antiphospholipid-Syndrom war, und ihre Antikoagulation wurde auf Warfarin umgestellt. Sie unterzog sich 16 Monate nach ihrer anfänglichen akuten Lungenembolie einer PEA am Royal Papworth Hospital, und die Nachsorge-RHC sechs Monate später bestätigte eine hämodynamische Verbesserung mit einem normalen mPAP von 23 mmHg, PAWP 11 mmHg und PVR 2,0 WU. Sildenafil wurde postoperativ abgesetzt. Die Patientin hatte anschließend eine erfolgreiche Schwangerschaft und setzt die lebenslange Antikoagulation fort.Abbildung 6 Beatmungs- und Perfusionsscan (V/Q) bei einem Patienten mit CTEPH (siehe Fall 3), der mehrere nicht übereinstimmende Perfusionsdefekte zeigt.
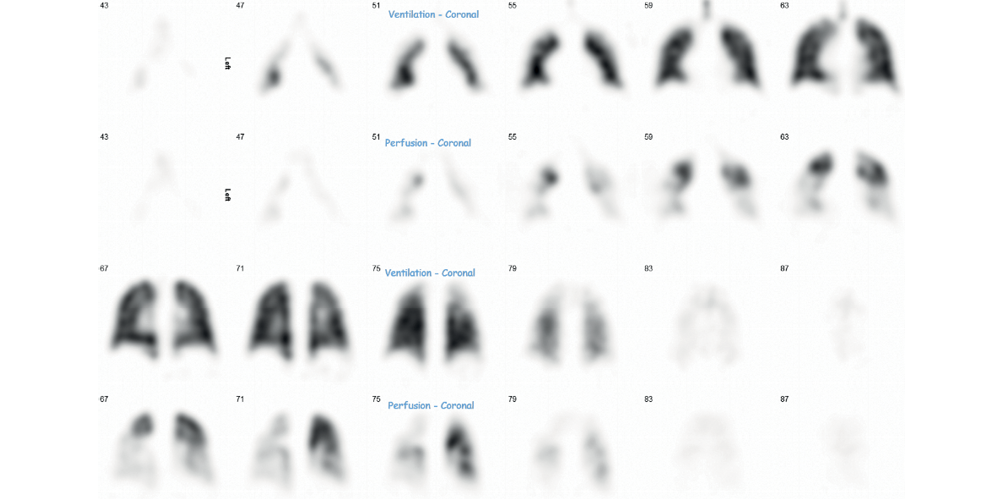
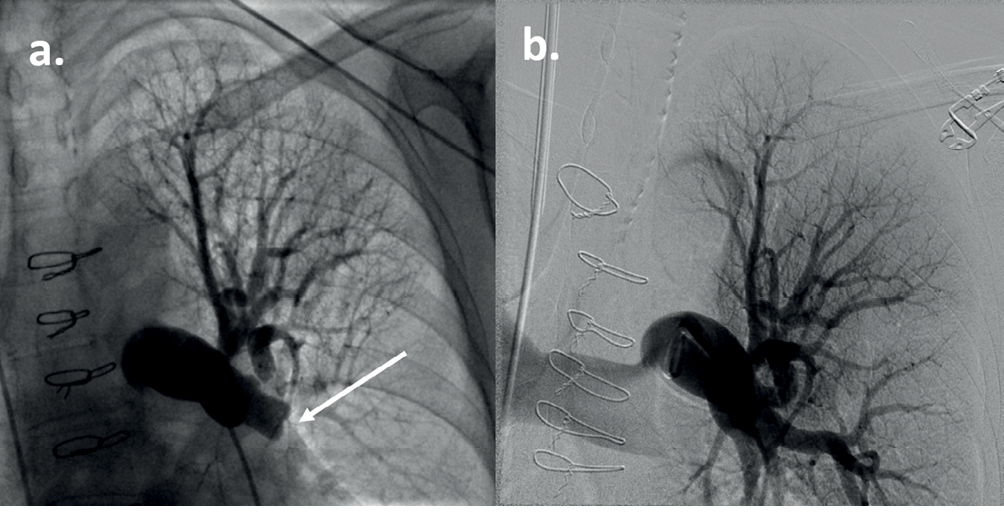
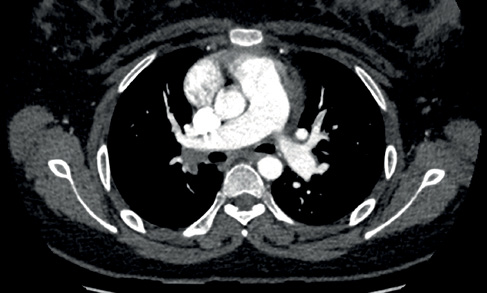
 Abbildung 7 CT-Lungenangiogramm eines Patienten mit CTEPH (siehe Fall 3). Es gibt Hinweise auf eine chronische thromboembolische Erkrankung der Lungenarterien. Die Hauptpulmonalarterie ist auf 34 mm dilatiert und breiter als die Aorta, was dem PH entspricht.
stubbs_figure_7.jpg
Abbildung 7 CT-Lungenangiogramm eines Patienten mit CTEPH (siehe Fall 3). Es gibt Hinweise auf eine chronische thromboembolische Erkrankung der Lungenarterien. Die Hauptpulmonalarterie ist auf 34 mm dilatiert und breiter als die Aorta, was dem PH entspricht.
stubbs_figure_7.jpg
 FazitPH ist eine vielschichtige Erkrankung, die aus einer Vielzahl von Grunderkrankungen resultiert. Die therapeutischen Möglichkeiten haben sich in den letzten zwei Jahrzehnten enorm erweitert, was zu erheblichen Verbesserungen der Prognose für einige Präsentationen geführt hat. Daher ist eine detaillierte Untersuchung unerlässlich, um die genaue Ätiologie und damit die optimale Behandlung zu ermitteln. Leider wird PH oft noch Monate oder Jahre nach dem Auftreten der Symptome diagnostiziert, und es ist unwahrscheinlich, dass Screening-Strategien dies in absehbarer Zukunft verbessern werden. Die Kenntnis der Ätiologie, des Erscheinungsbildes und der Behandlungsmöglichkeiten dieser Erkrankung ist daher in vielen Bereichen der Medizin wichtig, um die Behandlung dieser lebensverändernden Erkrankung zu verbessern.
FazitPH ist eine vielschichtige Erkrankung, die aus einer Vielzahl von Grunderkrankungen resultiert. Die therapeutischen Möglichkeiten haben sich in den letzten zwei Jahrzehnten enorm erweitert, was zu erheblichen Verbesserungen der Prognose für einige Präsentationen geführt hat. Daher ist eine detaillierte Untersuchung unerlässlich, um die genaue Ätiologie und damit die optimale Behandlung zu ermitteln. Leider wird PH oft noch Monate oder Jahre nach dem Auftreten der Symptome diagnostiziert, und es ist unwahrscheinlich, dass Screening-Strategien dies in absehbarer Zukunft verbessern werden. Die Kenntnis der Ätiologie, des Erscheinungsbildes und der Behandlungsmöglichkeiten dieser Erkrankung ist daher in vielen Bereichen der Medizin wichtig, um die Behandlung dieser lebensverändernden Erkrankung zu verbessern.
- Unspezifische frühe Symptome und Anzeichen
- Symptome und Anzeichen werden häufigeren Komorbiditäten zugeschrieben
- Seltener Zustand und daher ein niedriger Verdachtsindex
- Komplexer Präsentationsweg, der kardiologische, respiratorische oder andere Fachgebiete umfassen kann
- Mangelndes Bewusstsein für Überweisungswege, Richtlinien und tertiäre Zentren
- Keine wirksamen Screening-Strategien bei den meisten Erkrankungen, die PH verursachen, z. B. IPAH, chronische Herzerkrankungen, Lebererkrankungen
- Screening nicht durchgeführt, z. B. postpulmonale Embolie, Bindegewebserkrankung, genetisches Screening
- (A) Ventrikel: RV:LV Basaldurchmesser > 1,0, Abflachung des intraventrikulären Septums
- (
") Pulmonalarterie: Pulmonalklappenbeschleunigungszeit < 105 ms, Pulmonalarteriendurchmesser > 25 mm, frühdiastolische Lungeninsuffizienzgeschwindigkeit > 2,2 m/s
Pulmonalarterie: Pulmonalklappenbeschleunigungszeit < 105 ms, Pulmonalarteriendurchmesser > 25 mm, frühdiastolische Lungeninsuffizienzgeschwindigkeit > 2,2 m/s - (C) Untere Hohlvene und rechter Vorhof: verringerter inspiratorischer Kollaps der unteren Hohlvene, vergrößerter rechter Vorhof
OMNIA TEMPUS HABENT
Diagnose IPAH im Februar 2013, in Behandlung bei OA Dr. Ulrich Krüger, jetzt Dr. Fischer Herzzentrum Duisburg, Medikamente: Sildenafil, Bosentan jetzt Macitentan, Subkutane Treprostinilpumpe, seit Januar 2024 getunnelter ZVK mit externer Pumpe (Groshongkatheter), 24/7 Sauerstoff, Marcumar, Diuretika
Letzte Änderung: 13 Mai 2022 23:02 von danny.
Bitte Anmelden oder Registrieren um der Konversation beizutreten.